
Abstract
With congenital central hypoventilation syndrome (CCHS), most patients have a de novo 5–13 polyalanine expansion mutation in PHOX2B. We reported previously that de novo polyalanine expansion mutations were of paternal origin and were derived from unequal sister chromatid exchange during spermatogenesis in six and four informative families, respectively. In this study, we analyzed the relationship between haplotypes and de novo polyalanine expansion in PHOX2B and found that haplotypes carrying rs17884724:A>C were detected frequently in 7-alanine expanded (27-alanine) mutant alleles, which are the most prevalent mutations in CCHS. The allele with rs17884724:A>C made fewer nucleotide mismatches in the misalignment at crossing-over than the allele without rs17884724:A>C. The high frequency of rs17884724:A>C in 7-alanine expansion (27-alanine) mutations also supported the unequal crossover mechanism for polyalanine expansion. We also confirmed the paternal origin of de novo polyalanine expansion mutation and unequal sister chromatid exchange association in three more patients. In spite of paternal bias, the paternal age effect on CCHS incidence was not observed. De novo polyalanine expansion mutations are mainly derived from unequal sister chromatid exchange during spermatogenesis because of replication and/or repair systems that are specific for spermatogenesis.
Similar content being viewed by others


Introduction
Congenital central hypoventilation syndrome (CCHS; MIM 209880) is characterized by failure of the automatic control of breathing during sleep, and results from the dominant PHOX2B mutation. About 90% of patients have de novo polyalanine expansion mutations in the polyalanine tract of 20 residues.1, 2, 3, 4, 5, 6 Approximately 5% of patients inherit polyalanine expansion mutations mostly from asymptomatic parents with somatic mosaicism and rarely from affected parents. Polyalanine expansion disorders constitute one family of homopolymer expansion disorders, including at least nine disorders. Expanded polyalanine tracts are encoded for by imperfect GCA, GCG, GCC and GCT repeats; they are meiotically and somatically stable. Warren7 inferred unequal crossover as a causative mechanism of the polyalanine expansion of HOXD13 in synpolydactyly 1. In 2007, we reported that de novo polyalanine expansion mutations of PHOX2B were of paternal origin and were derived from unequal sister chromatid exchange in six and four informative CCHS families, respectively.8 In contrast, Trochet et al.9 suggested mechanisms other than unequal crossing-over on the basis of data of three rare complex polyalanine expansion mutations. Parodi et al.10 reported that de novo polyalanine expansion of PHOX2B was derived, respectively, from maternal allele and paternal allele in 7 and 13 CCHS patients.
In this paper, we studied the relationship between haplotypes and polyalanine expansion in PHOX2B and found that haplotypes carrying rs17884724:A>C are associated with a 7-alanine expansion (27-alanine) mutation, which supports that unequal crossover is involved in de novo polyalanine expansion mutation. We also confirmed paternal origin and association with unequal sister chromatid exchange during spermatogenesis of polyalanine expansion in three more CCHS patients.
Materials and methods
The ethics committee of the Yamagata University School of Medicine approved this study. After receiving written informed consent from the patients' families, peripheral blood was collected from patients and family members for genomic DNA extraction. We studied 39 patients (male/female ratio, 18:21) with CCHS, including 29 previously analyzed cases and 10 newly diagnosed patients.2, 8, 11 Seven patients carried 5, 13 patients carried 6, 14 patients carried 7, single patients each carried 10, 11 and 12, and 2 patients carried 13 polyalanine expansion mutations. We analyzed six single-nucleotide polymorphisms (SNPs) and three deletion–insertion polymorphisms (DIPs) in PHOX2B as follows: one DIP (rs72266779) and two SNPs (rs4608840 and rs6811325) in intron 1, one SNIP (rs2196822) and two DIPs (rs3038692 and rs10614480) in intron 2 and three SNPs (rs17884724, rs6826373 and rs11723860) in exon 3. They were all within the same haplotype block. For the analysis of SNPs and DIPs, we amplified the PHOX2B genome using three-set primers described in our previous paper.8 We studied all SNPs and DIPs by sequence determination using amplified products from genomic DNA and after subcloning of PCR-amplified products into the TA cloning vector. The primers used for sequence determination were designed on the basis of the genomic database (accession number NC_000004.10). We classified the haplotypes into five (A–E) groups on the basis of the information of SNPs and DIPs, and determined the parental origin and chromosomal events of polyalanine expansion by haplotype analysis. Paternity was confirmed using 16 markers provided with the AmpFLSTR identifier kit (PE Applied Biosystems, Foster City, CA, USA). Comparison of rs17884724:A>C frequencies in wild (20-alanine) alleles and mutant polyalanine expanded alleles was performed by Fisher's test. P-values <0.05 were considered to be significant.
Results
We detected polyalanine expansion mutations of PHOX2B in 39 patients. Familial mutant analysis and paternity testing indicated that each mutation in all families occurred as a de novo event.
To investigate the relationship between haplotypes and polyalanine expansion in PHOX2B, we determined six SNPs and three DIPs in 108 PHOX2Bs carrying the 20-alanine tract from healthy control individuals and 39 PHOX2Bs carrying the 25–33 (5–13 expanded) alanine tract from CCHS patients. On the basis of data from SNPs and DIPs, we classified them into five (A–E) haplotypes (Table 1). Table 2 presents a comparison of haplotypes between wild alleles with 20-alanine tracts and mutant alleles with 25 to 33-alanine tracts. The number of participants in our study limited the statistical analysis of the relationship between the haplotype and the polyalanine expansion. However, one allele of haplotype B and nine alleles of haplotype D were detected in 27-alanine (7-alanine expanded) mutant alleles, a most prevalent expansion mutation. Both haplotypes carry rs17884724:A>C. The frequency of rs17884724:A>C in the wild alleles with the 20-alanine tract was 0.10; however, the frequency of rs17884724:A>C in mutant alleles with the 27-alanine tract was 0.71, a significantly high frequency (P<0.01). In contrast, haplotypes B and D carrying rs17884724:A>C were very few (or none) in other polyalanine expansion mutations and there was no significant difference in the frequency of rs17884724:A>C between wild and other alanine expanded alleles. As presented in Figure 1, the allele with rs17884724:A>C is expected to form fewer nucleotide mismatches in the misalignment at crossover than the allele with rs17884724:A.

Inferred alignment of polyalanine tracts of PHOX2B producing 7-alanine expansion by unequal crossing-over. Each distinct alanine codon is represented as a unique circle. The allele with rs17884724:A>C is expected to form fewer nucleotide mismatches than the allele with rs17884724:A.
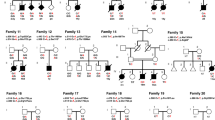
Regarding the paternal origin and alanine expansion mechanism, we studied 10 newly diagnosed families on the basis of haplotype information. We confirmed the paternal origin of expansion mutation and unequal sister chromatid exchange event during spermatogenesis in three informed families (Figure 2). We also identified three other patients who possibly received mutant alleles from their fathers because these patients inherited normal alleles from their mothers and were negative for the maternal disomy of chromosome 4 by the detection of paternal FGA (marker on chromosome 4) by a paternity test.

Parental origin of de novo polyalanine expansion. Affected individuals are represented as solid symbols. Haplotypes and length of polyalanine tracts are shown beneath each subject. Polyalanine expanded alleles are described in bold. The patient in family I had a mutant 27-alanine allele showing haplotype C, which was derived from the father. The patient in family II had a mutant 26-alanine allele showing haplotype C and the patient in family III had a mutant 25-alanine allele showing haplotype D; haplotype information revealed that each expanded allele was derived from the allele of each patient's father. The patients in families IV, V and VI inherited wild 20-alanine alleles from their mothers and had no maternal disomy of chromosome 4, suggesting that mutant polyalanine expanded alleles (27-alanine alleles in families IV and V, and 25-alanine allele in family VI) were transmitted from fathers.
To investigate whether the frequent mitotic division in paternal gametogenesis is associated with this paternal bias, we studied a ‘male age effect on the incidence of CCHS’ in 34 informative families. However, we were unable to detect any effect of paternal age on the offspring of patients, as depicted in Figure 3, suggesting that simple replication errors are not a major cause of de novo polyalanine expansion mutation of PHOX2B.

Paternal age effect on the incidence of congenital central hypoventilation syndrome (CCHS). Information regarding parental age was available for 34 of 39 patients. Black bars represent nine patients for whom paternal origin of polyalanine expansion was confirmed by haplotype analysis.
Discussion
Trinucleotide repeat sequences encoding polyalanine or polyglutamine tracts can form secondary structures. The formation of these secondary structures is considered to compromise DNA replication, thereby leading to expansion of those repeat sequences.12 In contrast to strand slippage in complete trinucleotide repeats encoding polyglutamine tracts in polyglutamine expansion disorders, incomplete trinucleotide repeats encoding polyalanine tracts in polyalanine expansion disorders were considered to result from an unequal crossover.7 As a matter of fact, CCHS is a polyalanine expansion disorder; most patients have a de novo 5–13 polyalanine expansion mutation in PHOX2B. In contrast, 5–13 polyalanine contractions are detected in normal healthy controls as polymorphisms. Combined with our previous report, we studied 23 families with a de novo polyalanine expansion and found that 9 (about 40%) families were informative for the parental origin of the mutant allele and all nine mutant alleles were of paternal origin. In addition, seven among nine (about 30%) families were also informative for chromosomal event and all seven mutations were derived from an unequal sister chromatid exchange during spermatogenesis.8 Regarding the relationship between haplotypes and polyalanine expansion, haplotypes carrying rs17884724:A>C were frequently detected in de novo 7-alanine expanded (27-alanine) alleles from unrelated patients. The 7-alanine expansion (27-alanine) mutation is the most prevalent expansion mutation. As presented in Figure 1, the allele with rs17884724:A>C is expected to form fewer nucleotide mismatches in the misalignment at crossover than the allele with rs17884724:A. Prevalence of rs17884724:A>C in 7-alanine expanded (27-alanine) alleles would also support that most de novo polyalanine expansion mutations are derived from an unequal crossover.
Regarding the parental origin and chromosomal events of polyalanine expansion, informative families by haplotype analysis are not many, about 30% of families. However, data from our informative families showed that all de novo polyalanine expansion mutations were of paternal origin and were derived from an unequal sister chromatid exchange during spermatogenesis.8 Parodi et al.10 studied 20 informative cases of parental origin by analyzing three SNPs and reported that de novo polyalanine expansion had occurred on the maternal allele in 7 patients and on the paternal allele in 13 patients. The reason for the difference from our data remains unclear; however, we cannot deny the possible influence of reaction artifacts during long PCR amplification. To avoid the influence of PCR errors, haplotype analysis seems to be a suitable method to study. Paternal expansion bias was also reported in most polyglutamine expansion disorders, another homopolymer expansion disorder,13 as well as in Duchenne muscular dystrophy and achondrodysplasia.14, 15 Frequent mitotic division in paternal gametogenesis might contribute to sex differences in polyalanine expansion mutation rates. However, a ‘male age effect’ was not observed, as depicted in Figure 3, suggesting that simple replication errors are not a major cause of de novo polyalanine expansion mutation of PHOX2B. Frequent unequal sister chromatid exchange was observed in the yeast with Sgs1 (homolog of human BLM) or Mph1 (homolog of human FANCM) mutants.16, 17 In addition, increased sister chromatid exchange frequency in Sgs1 mutants was reduced markedly by the disruption of the Rad52 or Msh2 gene, which is involved in mismatch repair.18 De Gregori et al.19 studied the parental origin of deletions in 5 de novo reciprocal translocations of chromosomes and 11 de novo complex chromosome rearrangements. They described that all were of paternal origin and spermatogenesis is more prone to generate multiple chaotic chromosome imbalances and reciprocal translocations than oogenesis. Replication and/or repair systems in spermatogenesis might differ from those in oocytogenesis and be associated with paternal bias of polyalanine expansion mutation.
There are a small number of asymptomatic or rarely symptomatic individuals with somatic mosaicism. Regarding the origin of polyalanine expansion in the somatic mosaicism, we studied the genome of an individual (a mother of a patient) with somatic mosaicism of 7-alanine expanded (27-alanine) and wild (20-alanine) alleles, but were unable to detect a 7-alanine contracted (13-alanine) allele, a counter allele of the expanded allele, which should have been produced at the recombination event (data not shown). Other groups also did not find a counter polyalanine contracted allele in the genome of individuals with somatic mosaicism.9, 10 Polyalanine expansion in somatic mosaicism is unlikely to have been derived from recombination. Trochet et al.9 reported three rare complex expansion mutations of PHOX2B and suggested that mechanisms other than an unequal crossing-over model would be involved. One of the three mutations is explained by recombination involving the codon encoding glycine 240 located 5′-upstream of the sequences encoding the polyalanine tract. However, two other complex expansion mutations are not explained by the recombination mechanism. Polyalanine expansion mutations in somatic mosaicism and few complex expansion mutations cannot be derived from unequal sister chromatid exchange, but are explainable by a replication mechanism such as a model of a repetitive hairpin formation on the nascent strand or a model of repeat instability generated during replication fork stalling and restart within the repetitive run.20
The results of our study suggest that unequal sister chromatid exchange during spermatogenesis is a major cause of de novo polyalanine expansion mutations, except for rare complex mutations and expansion mutations in somatic mosaicism.
Accession codes
References
Amiel, J., Laudier, B., Attie-Bitach, T., Trang, H., de Pontual, L., Gener, B. et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat. Genet. 33, 459–461 (2003).
Sasaki, A., Kanai, M., Kijima, K., Akaba, K., Hashimoto, M., Hasegawa, H. et al. Molecular analysis of congenital central hypoventilation syndrome. Hum. Genet. 114, 22–26 (2003).
Weese-Mayer, D. E., Berry-Kravis, E. M., Zhou, L., Maher, B. S., Silvestri, J. M., Curran, M. E. et al. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. Am. J. Med. Genet. A 123A, 267–278 (2003).
Matera, I., Bachetti, T., Puppo, F., Di Duca, M., Morandi, F., Casiraghi, G. M. et al. PHOX2B mutations and polyalanine expansions correlate with the severity of the respiratory phenotype and associated symptoms in both congenital and late onset central hypoventilation syndrome. J. Med. Genet. 41, 373–380 (2004).
Trochet, D., Hong, S. J., Lim, J. K., Brunet, J. F., Munnich, A., Kim, K. S. et al. Molecular consequences of PHOX2B missense, frameshift and alanine expansion mutations leading to autonomic dysfunction. Hum. Mol. Genet. 14, 3697–3708 (2005).
Berry-Kravis, E. M., Zhou, L., Rand, C. M. & Weese-Mayer, D. E. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am. J. Respir. Crit. Care Med. 174, 1139–1144 (2006).
Warren, S. T. Polyalanine expansion in synpolydactyly might result from unequal crossing-over of HOXD13. Science 275, 408–409 (1997).
Arai, H., Otagiri, T., Sasaki, A., Hashimoto, T., Umetsu, K., Tokunaga, K. et al. De novo polyalanine expansion of PHOX2B in congenital central hypoventilation syndrome: unequal sister chromatid exchange during paternal gametogenesis. J. Hum. Genet. 52, 921–925 (2007).
Trochet, D., de Pontual, L., Keren, B., Munnich, A., Vekemans, M., Lyonnet, S. et al. Polyalanine expansions might not result from unequal crossing-over. Hum. Mutat. 28, 1043–1044 (2007).
Parodi, S., Bachetti, T., Lantieri, F., Di Duca, M., Santamaria, G., Ottonello, G. et al. Parental origin and somatic mosaicism of PHOX2B mutations in congenital central hypoventilation syndrome. Hum. Mutat. 29, 206 (2008).
Horiuchi, H., Sasaki, A., Osawa, M., Kijima, K., Ino, Y., Matoba, R. et al. Sensitive detection of polyalanine expansions in PHOX2B by polymerase chain reaction using bisulfite-converted DNA. J. Mol. Diagn. 7, 638–640 (2005).
Wells, R. D., Dere, R., Hebert, M. L., Napierala, M. & Son, L. S. Advances in mechanisms of genetic instability related to hereditary neurological diseases. Nucleic Acids Res. 33, 3785–3798 (2005).
Yoon, S. R., Dubeau, L., de Young, M., Wexler, N. S. & Arnheim, N. Huntington disease expansion mutations in humans can occur before meiosis is completed. Proc. Natl Acad. Sci. USA 100, 8834–8838 (2003).
Hu, X. Y., Ray, P. N. & Worton, R. G. Mechanisms of tandem duplication in the Duchenne muscular dystrophy gene include both homologous and nonhomologous intrachromosomal recombination. EMBO J. 10, 2471–2477 (1991).
Crow, J. F. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 1, 40–47 (2000).
Ira, G., Malkova, A., Liberi, G., Foiani, M. & Haber, J. E. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115, 401–411 (2003).
Prakash, R ., Satory, D ., Dray, E ., Papusha, A ., Scheller, J., Kramer, W. et al. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 23, 67–79 (2009).
Onoda, F., Seki, M., Wang, W. & Enomoto, T. The hyper unequal sister chromatid recombination in an sgs1 mutant of budding yeast requires MSH2. DNA Repair (Amst) 3, 1355–1362 (2004).
De Gregori, M., Ciccone, R., Magini, P., Pramparo, T., Gimelli, S., Messa, J. et al. Cryptic deletions are a common finding in ‘balanced’ reciprocal and complex chromosome rearrangements: a study of 59 patients. J. Med. Genet. 44, 750–762 (2007).
Mirkin, S. M. Expandable DNA repeats and human disease. Nature 447, 932–940 (2007).
Acknowledgements
This study was partly supported by Grants-in-Aid for Scientific Research and by grants from the Ministry of Health, Labor, and Welfare. We thank H Izumino and Y Kishikawa for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Arai, H., Otagiri, T., Sasaki, A. et al. Polyalanine expansion of PHOX2B in congenital central hypoventilation syndrome: rs17884724:A>C is associated with 7-alanine expansion. J Hum Genet 55, 4–7 (2010). https://doi.org/10.1038/jhg.2009.109
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.109
Keywords
This article is cited by
-
Guidelines for diagnosis and management of congenital central hypoventilation syndrome
Orphanet Journal of Rare Diseases (2020)
-
Inheritance of polyalanine expansion mutation of PHOX2B in congenital central hypoventilation syndrome
Journal of Human Genetics (2012)
