
Abstract
Leptosphaerins H and I (1 and 2), two new xanthone derivatives, and six known compounds, leptosphaerin F (3), monodictysin B (4), norlichexanthone (5), leptosphaerin D (6), moniliphenone (7) and emodinbianthrone (8) have been isolated from a scale-up fermentation of the ascomycete fungus Leptosphaeria sp. Their structures were primarily elucidated by interpretation of NMR spectroscopic data. The absolute configuration of 1 was assigned using the modified Mosher method, whereas that of C-8a in 2 was determined via the CD data. Compound 6 showed modest cytotoxicity against a panel of three human tumor cell lines.
Similar content being viewed by others
Introduction
Fungi inhabiting special and competitive environments are more likely to produce bioactive secondary metabolites with diverse structural features, presumably due to their highly evolved metabolic systems adapted during the natural selection process.1, 2, 3, 4, 5 In recent years, more and more bioactive compounds from Cordyceps-colonizing fungi, the species that colonize the fruiting body of Cordyceps sinensis, and the fungi isolated from the soil samples in the C. sinensis, have been reported.6, 7, 8, 9 In the course of investigating new bioactive compounds from the fungus, a strain of Leptosphaeria sp. (XZC04-CS-304), isolated from the soil sample on the surface of the fruiting body of C. sinensis collected in Linzhi, Tibet, China was investigated, leading to the discovery of antifungal polyketides leptosphaerins A–G.10 Subsequent chemical investigations of the extract from a larger-scale fermentation of this fungus led to the isolation of two new xanthone derivatives, leptosphaerins H and I (1 and 2), together with the known compounds, monodictysin B (4),11 norlichexanthone (5),12 moniliphenone (7)13 and emodinbianthrone (8)14 (Figure 1). The previously identified polyketides, leptosphaerins F (3) and D (6) were also re-isolated in the current work. Details of the isolation, structure elucidation and biological activities of these compounds are reported herein.

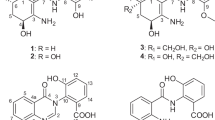
Chemical structures of compounds 1–8.
Results and discussion
Leptosphaerin H (1) was obtained as light yellow powder. The molecular formula was determined to be C15H16O4 (eight degrees of unsaturation) on the basis of its high-resolution electrospray ionisation mass spectrometry (HRESIMS) (m/z 283.0941 [M+Na]+; Δ+0.2 m.m.u.). The 1H and 13C NMR spectra of 1 (Table 1) revealed the presence of two methyl groups, one methylene unit, three methines (one oxygenated), eight aromatic/olefinic carbons (three oxygenated) and one α, β-unsaturated ketone carbon (δC 183.8). These data together with two exchangeable protons (δH 3.91, 12.9) accounted for all 1H and 13C resonances and required the presence of three rings for 1. Interpretation of the 1H–1H COSY NMR (Figure 2) data of 1 identified two isolated proton spin systems of C-2–C-4 and C-5–C-8 (including 7-OH, C-10 and C-11). Analysis of 1H NMR spectrum for 1 (Table 1) displayed the presence of one 1, 2, 3-trisubstituted benzene ring, which was also supported by HMBC correlations (Figure 2) from H-2 to C-1 and C-9a, H-3 to C-1 and C-4a, and from H-4 to C-4a. HMBC correlations from H-5 and H-8 to C-8a and C-10a, and from H3-11 to C-7, C-8 and C-8a indicated that the C-8a/C-10a olefin were attached to C-8 and C-5, respectively, establishing the cyclohexene ring. The cross peaks from OH-1 (δH 12.9) to C-1 and C-2 indicated that the hydroxyl group was attached to C-1. Further correlation from H-8 to C-9, together with the four-bond HMBC correlation from H-4 to C-9 implied that the C-9 was located between C-9a and C-8a. Considering the chemical shift values of C-4a (δC 157.1) and C-10a (δC 166.3), and the unsaturation requirement for 1, the two carbons were attached to the same oxygen to form tetrahydro-9H-xanthen-9-one skeleton. Collectively, these data permitted assignment of the planar structure of 1.

Key 1H–1H COSY, HMBC correlations for 1.
The relative configuration of 1 was assigned by analysis of the 1H–1H coupling constants and NOESY experimental data. The small coupling constant of between H-7 and H-8 (J=2.0 Hz) indicated that these two protons had a cis relationship with respect to the corresponding cyclohexene ring. NOESY correlations of H-6 with H3-11 placed H-6 and H3-11 on the same face of the ring system. Therefore, the relative configuration was proposed as shown.
The absolute configuration of 1 was determined using the modified Mosher method.15, 16 Treatment of 1 with (S)- and (R)-MTPA Cl alpha-Methoxy-alpha-(trifluoromethyl)phenylacetyl chloride afforded the major products, the R- (1a) and S-MTPA (1b) mono-esters, respectively. The difference in chemical shift values (Δδ=δS−δR) for the diastereomeric esters 1b and 1a was calculated to assign the 7S absolute configuration. Therefore, the 6R, 7S, 8R configuration was proposed for 1 on the basis of the Δδ results summarized in Figure 3.

Δδ Values (in p.p.m.)=δS−δR obtained for (S)- and (R)-MTPA esters 1b and 1a.
The molecular formula of leptosphaerin I (2) was deduced as C15H18O5 (seven degrees of unsaturation) by HRESIMS (m/z 301.1050 [M+Na]+; Δ−0.4 m.m.u.). A molecular formula search identified a tetrahydroxanthone, leptosphaerin F (3)10 and monodictysin B (4),11 which possess the same elemental composition as 2. Analysis of the NMR data of 2 (Table 2) revealed the same gross structure as 3 and 4, indicating their isomeric relationship. The relative configuration of 2 was assigned by analysis of its 1H-1H coupling constants and NOESY data. A large coupling constant of 10.1 Hz between H-5 and H-6 in 2 revealed their trans relationship. The small coupling constant 4.1 Hz between H-8a and H-8 in 2, which is similar with that in compound 4 (4.3 Hz),11 indicated that H-8a and H-8 were located on the same side of the cyclohexane-1,4-diol moiety. These assignments were confirmed by NOESY correlations (Figure 4) of H-6 with H-8 and H-8a. NOESY correlations of H3-11 with H3-12 revealed their proximity in space. Therefore, the relative configuration of 2 was proposed as shown.

Key NOESY correlations for 2.
The absolute configuration of C-8a in 2 was assigned by application of the CD exciton chirality method. The CD spectrum of 2 (Figure 5) showed a negative Cotton effect at 293 (Δɛ–0.3) and a positive Cotton effect at 368 (Δɛ+0.4) nm, respectively, similar to that of (−)-rotoic acid,17 suggesting 8aR absolute configuration. Considering the relative configuration determined by 1H-1H coupling constants and NOESY data, 2 was assigned 5R, 6S, 8R, 8aR and 10aS absolute configuration.

CD spectrum of 2 and 3. A full color version of this figure is available at The Journal of Antibiotics journal online.
The known compounds 3–8, isolated from the crude extract were identified as leptosphaerin F,10 monodictysin B,11 norlichexanthone,12 leptosphaerin D,10 moniliphenone13 and emodinbianthrone,14 respectively, by comparison of their NMR and MS data with those reported.
Compounds 1–8 were tested for cytotoxicity against three human tumor cell lines, HeLa (human cervical cancer), MCF-7 (human breast cancer) and HepG2 (human hepatocellular carcinoma). Compound 6 was cytotoxic to the three cell lines, showing IC50 values of 11.0, 14.7 and 11.0 μm, respectively, whereas the positive control 5-fluorouracil showed IC50 values of 10.0, 15.0 and 23.1 μm, respectively. Compounds 1–5, 7 and 8 did not show detectable inhibitory effects on the cell lines tested at 20 μg ml−1.
Chemical constituents of the fungus Leptosphaeria sp. have been studied to afford five new polyketides leptosphaerins A–G10 in our previous study. In the present study, chemical reinvestigation of Leptosphaeria sp. has led to the isolation of two additional new xanthone derivatives, leptosphaerins H and I (1 and 2). Compound 1 differs from the known 3, 4-dihydroglobosuxanthone A by having one more methyl group at C-6, and only a methyl group at C-8 instead of a hydroxyl group and acetyl in the latter.18 Compound 2 is a C-8 and C-10 a stereoisomer of leptosphaerin F.10 Biogenetically, compounds 1–8 could be derived from the cleavage of an anthraquinone/anthrone precursor.19, 20
Methods
General experimental procedures
Optical rotations were measured on a Perkin-Elmer 241 polarimeter (PerkinElmer Waltham, MA, USA), and UV data were recorded on a Shimadzu Biospec-1601 spectrophotometer (Shimadzu, Columbia, MD, USA). CD spectra were recorded on a JASCO J-815 spectropolarimeter (Jasco, Tokyo, Japan) using MeOH as solvent. IR data were recorded using a Nicolet Magna-IR 750 spectrophotometer. 1H and 13C NMR data were acquired with Varian Mercury -500 and -600 spectrometers (Varian, Inc., Palo Alto, CA, USA) using solvent signals (acetone-d6; δH 2.05/δC 29.8, 206.1) as references. The HMQC and HMBC experiments were optimized for 145.0 and 8.0 Hz, respectively. ESIMS data were recorded on a Bruker Esquire 3000plus spectrometer (Bruker, Bremen, Germany), and HRESIMS data were obtained using Bruker APEX III 7.0 T and APEXII FT-ICR spectrometers (Bruker Daltonics, Billerica, MA, USA), respectively.
Fungal material
The ascomycete fungus Leptosphaeria sp. (XZC04-CS-304) was isolated by Dr Mu Wang from the soil sample on the surface of the fruiting body of C. sinensis collected in Linzhi, Tibet, China. A fresh sample of C. sinensis was collected, together with the soil partially covered on the surface of its fruiting body, and sealed in a plastic bag. The strain was isolated from the soil suspension in distilled water by the spread-plate technique on a potato dextrose agar plate with streptomycin. The isolate was identified by one of the authors and assigned the accession number XZC04-CS–304 in X.L.’s culture collection at the Institute of Microbiology, Chinese Academy of Sciences, Beijing. The fungal strain was cultured on slants of potato dextrose agar at 25 °C for 10 days. Agar plugs were used to inoculate in 250 ml Erlenmeyer flasks, each containing 50 ml of media (0.4% glucose, 1% malt extract and 0.4% yeast extract), and the final pH of the media was adjusted to 6.5 before sterilization. Flask cultures were incubated at 25 °C on a rotary shaker at 170 r.p.m. for 5 days. Fermentation was carried out in 12 Fernbach flasks (500 ml), each containing 80 g of rice. Spore inoculum was prepared by suspension in sterile, distilled H2O to give a final spore/cell suspension of 1 × 106 ml−1. Distilled H2O (100 ml) was added to each flask, and the contents were soaked overnight before autoclaving at 15 lb/in.2 for 30 min. After cooling to room temperature, each flask was inoculated with 5.0 ml of the spore inoculum and incubated at 25 °C for 40 days.
Extraction and isolation
The fermented material was extracted repeatedly with ethyl acetate (4 × 500 ml), and the organic solvent was evaporated to dryness under vacuum to afford the crude extract (8.2 g), which was fractionated by silica gel VLC using petroleum ether–EtOAc gradient elution. The fraction (286 mg) eluted with 45% EtOAc was separated by Sephadex LH-20 column chromatography eluting with 1:1 CH2Cl2–MeOH. The resulting subfractions were combined and further purified by reverse phase HPLC (Agilent Zorbax SB-C18 column (Agilent Technologies, Wilmington, DE, USA); 5 μm; 9.4 × 250 mm; 20% MeOH in H2O for 2 min, from 20 to 65% in 30 min; 2 ml min−1) to afford 5 (10.7 mg, tR 21.9 min) and 7 (25.0 mg, tR 27.5 min). The fraction (90 mg) eluted with 25% EtOAc was purified by RP HPLC (25% MeOH in H2O for 5 min, from 25 to 80% in 40 min) to afford 1 (6.9 mg, tR 16.6 min), 2 (8.2 mg, tR 25.1 min), 3 (7.4 mg, tR 25.2 min) and 4 (10.0 mg, tR 25.4 min). The fraction (90 mg) eluted with 50% EtOAc was also separated by RP HPLC (60% MeOH in H2O for 5 min, from 80 to 90% in 25 min) to afford 6 (6.9 mg, tR 22.5 min) and 8 (28.1 mg, tR 24.2 min).
Leptosphaerin H (1)
Light yellow powder; [α]25D –33.3 (c 0.1, MeOH); UV (MeOH) λmax (log ɛ) 237 (3.79), 330 (3.03) nm; IR (neat) νmax 3482, 3451, 2969, 2933, 1648, 1616, 1587, 1487, 1362, 1270, 1234, 986, 775 cm−1; 1H NMR, 13C NMR, HMBC and NOESY data see Table 1; HRESIMS m/z 283.0941 (calcd for C15H16O4Na, 283.0943).
Preparation of (R)-MTPA ester (1a) and (S)-MTPA ester (1b)
A sample of 1 (1.0 mg, 0.004 mmol), (S)-MTPA Cl (2.0 μl, 0.011 mmol) and pyridine-d5 (0.5 ml) was allowed to react in an NMR tube at ambient temperature for 24 h. The 1H NMR data of the R-MTPA ester derivative (1a) were obtained directly on the reaction mixture: 1H NMR (CDCl3, 500 MHz) δ 7.50 (1H, t, 8.0 Hz, H-3), 6.80 (1H, d, 8.0 Hz, H-2), 6.78 (1H, d, 8.0 Hz, H-4), 3.31 (1H, dq, 2.0, 7.1 Hz, H-8), 2.65 (1H, dd, 4.3, 11.4 Hz, H-5a), 2.44 (1H, dd, 11.4, 16.6 Hz, H-5b), 2.40 (1H, m, H-6), 1.30 (3H, d, 7.1 Hz, H3-11), 1.18 (3H, d, 7, H3-10).
Another sample of 1 (1.0 mg, 0.004 mmol), (R)-MTPA Cl (2.0 μl, 0.011 mmol), and pyridine-d5 (0.5 ml) was processed as described above for 1a to afford 1b: 1H NMR (CDCl3, 500 MHz) δ 7.52 (1H, t, 8.0 Hz, H-3), 6.84 (1H, d, 8.0 Hz, H-2), 6.82 (1H, d, 8.0 Hz, H-4), 3.37 (1H, dq, 2.0, 7.1 Hz, H-8), 2.66 (1H, dd, 4.3, 11.4 Hz, H-5a), 2.43 (1H, dd, 11.4, 16.6 Hz, H-5b), 2.39 (1H, m, H-6), 1.32 (3H, d, 7.1 Hz, H3-11), 1.09 (3H, d, 7, H3-10).
Leptosphaerin I (2)
White solid; [α]25D –81.5 (c 0.1, CH3OH); UV (MeOH) λmax (log ɛ) 218 (3.93), 277 (3.78), 353 (3.28) nm; CD (c 1.0 × 10−4 m, CH3OH) λmax (Δɛ) 293 (–0.3), 368 (+0.4) nm; IR (neat) νmax 3460, 2975, 2920, 1614, 1460, 1358, 1229, 1044, 797, 768, 731 cm−1; 1H and 13C NMR data see Table 2; HRESIMS m/z 301.1050 (calcd for C15H18O5Na, 301.1046).
Leptosphaerin F (3)
1H, 13C NMR and the MS data were consistent with literature values.10
Monodictysin B (4)
1H, 13C NMR and the MS data were consistent with literature values.11
Norlichexanthone (5)
1H, 13C NMR and the MS data were consistent with literature values.12
Leptosphaerin D (6)
1H, 13C NMR and the MS data were consistent with literature values.10
Moniliphenone (7)
1H, 13C NMR and the MS data were consistent with literature values.13
Emodinbianthrone (8)
1H, 13C NMR and the MS data were consistent with literature values.14
MTT assay
The assay was run in triplicate.21 In 96-well plates, each well was plated with 104 cells. After cell attachment overnight, the medium was removed, and each well was treated with 50 μl of medium containing 0.2% DMSO, or appropriate concentrations of the test compounds and positive control 5-fluorouracil (10 mg ml−1 as stock solution of a compound in DMSO and serial dilutions; the test compounds showed good solubility in DMSO and did not precipitate when added to the cells). Cells were treated at 37 °C for 4 h in a humidified incubator at 5% CO2 first, and were allowed to grow for another 48 h after the medium was changed to fresh Dulbecco's Modified Eagle Medium (DMEM). MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) (Sigma, St Louis, MO, USA) was dissolved in serum-free medium or phosphate-buffered saline at 0.5 mg ml−1 and sonicated briefly. In the dark, 50 μl of MTT/medium was added into each well after the medium was removed from wells, and incubated at 37 °C for 3 h. Upon removal of MTT/medium, 100 μl of DMSO was added to each well, and agitated at 60 r.p.m. for 5 min to dissolve the precipitate. The assay plate was read at 540 nm using a microplate reader.
References
Gloer, J. B. Antiinsectan natural products from fungal sclerotia. Acc. Chem. Res. 28, 343–350 (2002).
Macíasrubalcava, M. L., Sobrino, R. V., Meléndezgonzález, C. & Hernándezortega, S. Naphthoquinone spiroketals and organic extracts from the endophytic fungus edenia gomezpompae as potential herbicides. J. Agric. Food Chem. 62, 3553–3562 (2014).
Keller, N. P. & Wiemann, P. Strategies for mining fungal natural products. J. Ind. Microbiol. Biotechnol. 41, 301–313 (2014).
Tan, R. X. & Zou, W. X. Endophytes: a rich source of functional metabolites. Nat. Prod. Rep. 18, 448–459 (2001).
Dreyfuss, M. M. & Chapela, I. H. CHAPTER 3–Potential of fungi in the discovery of novel, low-molecular weight pharmaceuticals. Biotechnology 26, 49–80 (1994).
Ma, C. et al. N-hydroxypyridones, phenylhydrazones, and a quinazolinone from Isaria farinosa. J. Nat. Prod. 74, 32–37 (2011).
Zhang, Y., Liu, S., Che, Y. & Liu, X. & Epicoccins, A-D Epipolythiodioxopiperazines from a Cordyceps-colonizing isolate of Epicoccum nigrum. J. Nat. Prod. 70, 1522–1525 (2007).
Guo, H. et al. Bioactive p-terphenyl derivatives from a Cordyceps-colonizing isolate of Gliocladium sp. J. Nat. Prod. 70, 1519–1521 (2007).
Lin, J. et al. Isolation and characterization of aphidicolin and chlamydosporol derivatives from t olypocladium inflatum. J. Nat. Prod. 74, 1798–1804 (2011).
Lin, J. et al. Polyketides from the ascomycete fungus Leptosphaeria sp. J. Nat. Prod. 73, 905–910 (2010).
Krick, A. et al. Potential cancer chemopreventive in vitro activities of monomeric xanthone derivatives from the marine algicolous fungus Monodictys putredinis. J. Nat. Prod. 70, 353–360 (2007).
Mutanyatta, J., Matapa, B. G., Shushu, D. D. & Abegaz, B. M. Homoisoflavonoids and xanthones from the tubers of wild and in vitro regenerated Ledebouria graminifolia and cytotoxic activities of some of the homoisoflavonoids. Phytochemistry 62, 797–804 (2003).
Kachi, H., Sassa, T., Kachi, H. & Sassa, T. Isolation of monilphenone, a key intermediate in xanthone biosynthesis from Monilinia fructicola. Agric. Biol. Chem. 50, 1669–1671 (1986).
Abegaz, B. M. & Peter, M. G. Emodin and emodinanthrone rhamnoside acetates from fruits of Rhamnus prinoides. Phytochemistry 39, 1411–1414 (1995).
Dale, J. A. & Mosher, H. S. Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and. alpha.-methoxy-.alpha.-trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc. 95, 512–519 (1973).
Ohtani, I., Kusumi, T., Kashman, Y. & Kakisawa, H. A new aspect of the high-field NMR application of Mosher's method. The absolute configuration of marine triterpene sipholenol A. J.Org.Chem. 56, 1296–1298 (1991).
Lawson, M. A., Kaouadji, M., Allais, D. P., Champavier, Y. & Chulia, A. J. Substituted tubaic acids, new oxidative rotenoid metabolites from Lonchocarpus nicou. Tetrahedron Lett. 47, 451–454 (2006).
Karsten, K. et al. Xanthones and oxepino[2, 3-]chromones from three endophytic fungi. Chemistry 15, 12121–12132 (2009).
Turner, W. B. & Aldridge, D. C. Fungal Metabolites II 156–167 (Academic Press, New York, USA, 1983)..
Chexal, K. K., Holker, J. S. E. & Simpson, T. J. ChemInform abstract: the biosynthesis of fungal metabolites. Part V. Structure of variecoxanthones A, B, and C, metabolites of Aspergillus variecolor. J. Chem. Soc. Perkin 1 6, 549–554 (1975).
Koparal, A. T., Tüylü, B. A. & Türk, H. In vitro cytotoxic activities of (+)-usnic acid and (-)-usnic acid on V79, A549, and human lymphocyte cells and their non-genotoxicity on human lymphocytes. Nat. Prod. Res. 20, 1300–1307 (2006).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21372004, 21302216), the Program of the Excellent Young Scientists of Chinese Academy of Sciences and Natural Science Fund of Colleges and Universities in Jiangsu Province (16KJA180005, 16KJB180006).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Lin, J., Wang, R., Xu, G. et al. Two new polyketides from the ascomycete fungus Leptosphaeria sp.. J Antibiot 70, 743–746 (2017). https://doi.org/10.1038/ja.2017.5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2017.5
This article is cited by
-
Synthesis and antibacterial activity of novel lincomycin derivatives. IV. Optimization of an N-6 substituent
The Journal of Antibiotics (2017)
