
Abstract
Tacrolimus is an immunosuppressant macrolactam of fermentative origin. By means of HPLC, LC-MS and NMR analyses, coupled with the reference standard synthesis, the main impurities of tacrolimus bulk drug samples were identified and their chemical-physical properties reported. Known ascomycin and tautomers I and II were detected. The correct relative retention time HPLC value of 39,40-dihydro tacrolimus was established. The not described 23,24-anhydro derivative was detected and completely characterized. A full characterization of ascomycin and 39,40-dihydro tacrolimus was also reported.
Similar content being viewed by others
Introduction
Tacrolimus 1 is an immunosuppressant 23-membered macrolactam produced by Streptomyces tsukubaensis and used for the prevention of organ rejection following transplantation.1 The restricted rotation of the amide bond leads to the existence in solution of two conformational isomers (rotamers); the ratio of these rotamers is solvent-dependent and they are detectable by low-column temperature HPLC2 and 1H NMR.3
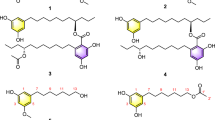
In addition, the presence of three adjacent carbonyl groups is responsible for the formation of two tautomeric structures, identified after isolation and purification in 1993:2 one lacking the hemiketal ring, compound 2 (tautomeric compound I), and the other having opposite stereochemistry at C-10, compound 3 (tautomeric compound II). (see Figure 1).

Structures of tacrolimus and related compounds.
We decided to study the impurity profile of 1 by analyzing six samples, five produced by fermentation in our plant and a commercial sample, by means of a LC-MS method. This LC method (reverse phase (RP) ‘in-house’ method in Table 1) was previously developed and utilized in order to monitor the progress of the fermentation.
Because at present there is no existing International Pharmacopoeia monograph, impurity profile of 1 is a topic of the US Pharmacopoeial Forum (PF)4, 5 that has listed tacrolimus-related by-products and equilibrium compounds (the structures and the adopted skeleton numbering convention are reported in Figure 1) and proposed their limits (see Table 1) by means of a normal phase HPLC method (method A). Tautomers 2 and 3, listed in the PF,4 were not included among the specified impurities being equilibrium compounds directly related to the drug substance.
On the basis of our LC-MS data, we proposed the structures for the detectable related by-products and the same samples were further analyzed by means of the analytical HPLC/UV PF method4 (method A) to test our hypothesis. By comparison of the observed relative retention time (RRt) values with the published data (see Table 1), we detected tautomers I and II in all the examined samples, the so called ‘diene’ and ‘compound A’ and, in addition, an impurity with RRt 0.87 not reported in the PF (see Table 1). On the contrary, the proposed aldehyde 44 was never detected, nor was a recently reported 18-methyl derivative.6 In a 2010 ‘in-process revision’ by the PF,5 a second, RP HPLC method (method B) (not applicable as such to a LC-MS analysis due to the presence in the mobile phase of non-volatile compounds) was proposed in order to overcome some inconsistencies unveiled, as suggested also by our group to the PF, in the previous method. In fact, two compounds (ascomycin 6 and dihydro derivative 7) showing the same RRt value (in method A) cannot be individually quantified. Following this second method, the presence of dihydro derivative 7 was detected (Table 1). We planned the synthesis of anhydrotacrolimus 5 and dihydro derivative 7, both not commercially available and still not characterized, necessary as the reference standards for a spiked HPLC analysis with authentic, well-characterized compounds.
Results
The preliminary LC-MS analytical investigation was carried out on five crude samples of fermentation broth that, containing major amounts of by-products, could furnish more information.
The results of LC-MS analysis showed the presence of two tacrolimus isomers (826.5 m z−1 corresponding to [M+Na]+) and three other compounds with 814.8, 828.5 and 809.0 m z−1, respectively. These peaks could be assigned, in principle, to ascomycin 6, dihydrotacrolimus 7 and anhydrotracrolimus 5, as sodium adducts. Because the five samples showed a comparable impurity profile, we decided to proceed with the analytical work on one of these samples and on a commercially available sample extracted from Astellas’ Prograf. Following the HPLC method (normal phase) described in the US PF,4 we detected at the reported RRt values the presence of tautomers I and II and the presence of derivative 5. The possible presence of ascomycin 6 and propyl derivative 7 was questionable because both the compounds were described with the same RRt value (0.96). On the basis of our previous LC-MS study, we could suggest the presence of both compounds. Moreover, using the HPLC method of PF,4 we detected the presence of an unknown peak, not reported in PF, with RRt 0.87. Two related compounds 5 and 7 were thus synthesized to determine if one of them might be RRt 0.87. In fact, LC-MS analysis showed the presence of compounds with 809.0 and 828.5 m z−1, probably corresponding to compounds 5 and 7, respectively.
Additionally, authentic ascomycin 6 was already available from internal source, because it was utilized in the past as starting material for the pimecrolimus synthesis.7
23,24-anhydro tacrolimus 5 was prepared, in 27% yield, by treatment of 1 with p-toluensulfonic acid in toluene at reflux, similarly to a described synthesis of 23,24-anhydro ascomycin.8 A characteristic vicinal NMR coupling constant (J) value (16 Hz) of H-23 and H-24 at 6.35 and 7.09 p.p.m., respectively, (see Table 2) was accounted for a E configuration of the 23,24 double bond of synthesized 5. Because chemical-physical properties of 5 were not reported, it was characterized by means of 1H NMR, MS and FT-IR analyses and the results are collected in Tables 2 and 3.
Required reference compound 7 was then prepared by a 5% Pd/C catalyzed hydrogenation of terminal 39,40 double bond of 1, selectively reduced with respect to the two trisubstituted double bonds present in the molecule, as suggested by 1H NMR spectrum: in fact, the multiplets at 5.82 and 5.85, due to the proton at position 39 of two rotamers of tacrolimus, were absent (see Table 2). The propyl derivative 7 is only cited, but not described, in the work of 1987 that was aimed at elucidating the structure of 1.3 A complete characterization, as for compound 5, was required, and chemical-physical properties and 1H NMR of 7 are collected in Tables 2 and 3.
The HPLC analysis of fermentation-broth sample, spiked with the reference standards, confirmed the presence of 23,24-anhydro derivative 5 and ascomycin 6 and furnished the proof that the impurity with RRt 0.87 corresponds to the propyl derivative 7; the peak with RRt 0.96 is only due to ascomycin 6. These results were further confirmed utilizing the HPLC method, more recently proposed (June 2010) by the PF.5 In this monograph proposal, the related by-products 5–7 were identified and quantified by means of a RP HPLC method. On the basis of these data, we can conclude that both the proposed HPLC methods are able to discriminate between ascomycin 6 and propyl derivative 7; the same 0.96 value reported for ascomycin 6 and propyl derivative 7 is a mistake (probably typing) because in our hands the first PF method shows a RRt 0.87 for compound 7.
The main difference between the two PF analytical methods refers to the quantization of the tautomeric forms I and II: in fact, the presence of water in the second HPLC method5 does not allow the correct evaluation of their amount in the examined solid sample. It is known that the composition of the mixture at equilibrium among tacrolimus and tautomers I and II varies with the water content.2, 9 Moreover, among the listed related by-products, detected with this RP HPLC method, the ‘anhydro’ derivative is not reported; using the prepared reference standard of anhydro derivative 5, we have verified that in these analytical conditions this compound is eluted at RRt 1.68. The two RP HPLC methods (method B and ‘in-house method’) offer the advantage to be utilized directly on the broths of fermentation, whereas the normal phase method requires, for the analysis of aqueous samples, a careful extraction procedure.
Because the studied tacrolimus sample did not show the desired purity (see Table 4), a purification process was required. In fact, ascomycin 6, as well as compounds 5 and 7, was present in tacrolimus crude sample in amounts (2.3%, 1.1% and 4.2%, respectively) exceeding the limits of specifications (see Table 4). After a filtration on an adsorption resin (XAD), a traditional column chromatography on silica gel (37:1 w/w) impregnated with silver nitrate (5% of stationary phase) was performed. Isocratic elution with n-heptane/ethyl acetate 3:7 afforded, in order, propyl derivative 7 and diene 5 always mixed with ascomycin 6, and then pure ascomycin followed by tacrolimus 1. Final crystallization provided compound 1 (sample A in Table 1) with all the specified impurities within the required limits and 40% overall yields from crude tacrolimus. Also, the remaining four crude samples were submitted to the same purification process, affording comparable results (samples B–E in Table 4). The HPLC data are reported in Table 4, related to our five samples and to commercial sample, obtained according to the PF method A, obviously taking into account the correct RRt value of propyl derivative 7. In our hands, the silver impregnated stationary phase has been reutilized three times without significant loss of efficiency. Other purification examples of crude tacrolimus by means of chromatographies in presence of silver ions are reported; for example, a resin10 was used as stationary phase or a high-speed counter current chromatography11 was carried out. We preferred the traditional more simple method of separation on impregnated silica gel affording satisfactory results on a multi-kg scale either as final sample purity or yields.
NMR study of compounds 1 and 5–7
An accurate NMR study of tacrolimus 1 and its related compounds 5–7 was done through 1D and 2D experiments.The spectra, initially recorded in deuterated chloroform following the literature data reported for 1 and 6,12, 13 or deuterated methanol, were finally done in deuterated pyridine (Pyd5), solvent that allowed a better spread of the proton resonances for all the studied compounds. In particular, all the methyl protons resulted well resolved and two sets of signals were identified in a 65:35 ratio due to the presence of two rotamers. A direct comparison of selected 1H NMR resonances of 1 and 5–7 in Pyd5 is showed in Table 2.
The study was focused on the signals of that part of the macrolactam system that makes each compound different from the others, and the strategy used for resonances assignment was the same for all the compounds. In particular, in the case of tacrolimus 1 and its 23,24-anhydro derivative 5, the characteristic geminal protons linked to the unsaturated C-40 (about 5 p.p.m., see Table 2) were used as entry points to assign 39, 38, 21 and 20 protons (see Table 2). COSY cross-peaks due to the allylic coupling of H-20 with H-18 and methyl-37 (singlet) allowed to identify also H-17 and methyl-36 (doublet). To complete the assignments, the characteristic singlet of methyl-42 (about 1.8 p.p.m., see Table 2) was used as second entry point. Its allylic coupling allowed to determine H-28 and subsequently H-29 signals. The second allylic coupling of H-28 was then used to assign H-26 and, from this resonance, H-25, H-24, H-23 and methyl-41 (doublet) were assigned. In the same way, but starting from the characteristic methyl-39 triplet of ascomycin 6 or methyl-40 triplet of the 21-propylderivative 7, the resonances of these compounds were also assigned (see Table 2).
Discussion
The reported study has allowed to achieve an unequivocal impurity profile characterization of tacrolimus produced by S. tsukubaensis fermentation, confirming the presence of the reported compounds not only by comparison with the sometime confusing reported RRt values4 but also by spiked analyses with authentic samples of detected impurities and by a LC-MS investigation. In addition, availability of compound 7 allowed to establish its correct RRt value (that is, 0.87 and not 0.96, using the PF method A4). Compounds 5 and 7, not yet characterized, were prepared and their chemical-physical properties reported. A careful NMR study in deuterated pyridine (Pyd5), which confirmed the proposed structures, was also performed comparing compounds 5–7 with tacrolimus 1. The suitable selection of the chromatographic fractions eluted from the silver nitrate-impregnated silica gel column (on 1–2 kg scale) was realized by means of a HPLC analysis (‘in-house’ method). The column chromatography purification followed by the final crystallization gave us the possibility to lower the initial amount of compounds 5–7 to the desired values (see Table 4).
The RP HPLC methods are suitable for the analysis of crude samples and their aqueous solution without affecting the performance of the HPLC column while the normal phase HPLC method may be utilized only on purified samples.
Because tacrolimus is of fermentative origin, the presence of ascomycin 6, an antifungal antibiotic isolated from Streptomyces hygroscopicus var. ascomyceticus by Arai et al.14 in 1962, can be explained considering that both the macrolactams are produced by strains of Streptomyces. It is questionable if compounds 2 and 3 had to be considered with a specific limit because they are tautomeric forms of the drug substance. Moreover, their relative quantization requires a normal phase HPLC method, because the presence of water affects the composition of the mixture at equilibrium. Dihydro derivative 7 is a reported, even if not characterized, fermentation product,15, 16 whereas 23,24-anhydro derivative 5 is a degradation product of tacrolimus: this impurity may be formed during the downstream process by thermal treatment in acid conditions (pasteurization).
Methods
Sample origin and preparation
Crude samples: tacrolimus-producing strain of S. tsukubaensis has been developed from the original strain S. tsukubaensis 9993 Ferm BP 927 through chemical and physical mutagen treatments performed in EUTICALS SpA (Milano, Italy). Industrial inoculums are prepared by seeding a previously sterilized media with slants of S. tsukubaensis. The growth of the microorganism is performed in laboratory incubator (about 1100, ml of seeded media in 5000, ml flasks at the temperature of 28 °C (±2 °C) at 120 r.p.m.) and takes 20–25 h. Then the inoculum is transferred to the fermentation plant for the vegetative step (growth of the mycelium). The mycelium suspension obtained at the end on the vegetative growth step (27±2 °C; under stirring; about 30% of dissolved oxygen) was fed in the fermentation vessel: the fermentation phase takes 10 days. In this phase, the dextrin concentration is maintained at a value >30 g l−1 by the addition of corn dextrin. The composition of the medias (vegetative and fermentation medias have the same composition) include: calcium carbonate (0.1–0.5 g l−1), corn dextrin (30–100 g l−1), dextrose (5.5 g l−1), glycerine (10 g l−1), iron sulphate (0.5 g l−1), dried yeast (10 g l−1) and continuous pH control (target value 6.6–6.9 maintained by addition of 30% sodium hydroxide). The examined crude tacrolimus samples were prepared by extraction with ethyl acetate (3 × 10 ml) of the broth (10 ml) after 10 days of fermentation. The combined organic phases were dried on sodium sulfate, filtered and evaporated under vacuum to afford crude tacrolimus samples. Five industrial fermentation broths samples, prepared as described above, were examined. Purified samples were obtained according to the procedure (silver nitrate-impregnated silica gel chromatography) described in the experimental section. Prograf sample: the collected content of 20 capsules was suspended in water (15 ml), the aqueous phase was extracted with ethyl acetate (3 × 15 ml) and the organic phase was treated as above.
HPLC
The HPLC system consisted of an Agilent 1100-series liquid chromatograph (Agilent, Santa Clara, CA, USA), equipped with auto-injector, diode array detector and a Chemstation software installed on a PC for data collecting and processing. ‘In-house’ method. A symmetry C18 (Waters, Milford, MA, USA) column (100 × 4.6 mm i.d., 3.5 μm) was employed in isocratic mode (0.1% acetic acid:acetonitrile:tetrahydrofuran 58:12:27 in volume) at a flow rate of 1.0 ml min−1 at 50 °C. Detector wavelength was set at 220 nm. A total of 10 μl of a sample solution in methanol (1 mg ml−1) was injected. The US PF conditions. Method A. A Supelcosil LC-diol (Supelco, Bellefonte, PA, USA) column (250 × 4.6 mm i.d., 5 μm) was employed in isocratic mode (n-hexane:n-butyl chloride:acetonitrile 7:2:1 in volume), at a flow rate of 1.5 ml min−1, at room temperature. Detector wavelength was set at 225 nm. A total of 20 μl of a sample solution in mobile phase (2 mg ml−1) were injected. Method B. A Luna C18 (150 mmx4.6 mmi.d., 3 μm) was employed in gradient mode (solution A: 6 mM phosphoric acid; solution B: acetonitrile:tert-butylether 81:19 v/v; solution C: solution A:solution B 4:1 v/v; solution D: solution A:solution B 1:4 v/v; Gradient program: 30 min C:D 72:28 v/v, 23 min C:D 15:85 v/v, 7 min C:D 72:28 v/v) at a flow rate of 1.5 ml min−1 at 60 °C. Detector wavelength was set at 220 nm. A total of 20 μl of a sample solution in acetonitrile:water 7:3 v/v (3 mg ml−1) was injected.
TLC
TLC analyses were performed on Silica Gel 60 F254 precoated plates with a fluorescent indicator (Merck, Darmstadt, Germany) with detection by spraying with 10% phosphomolybdic acid ethanol solution and heating at 110 °C.
Column chromatography
Column chromatography of compound 5 was carried out using silica gel 60 (70–230 mesh) (Merck). Purification of 1 was performed with the same silica gel impregnated with silver nitrate.
Mass spectrometry
Mass spectra were recorded on an Agilent instrument (Mod. 6330 Ion trap LC/MS) using the ESI source with positive ion polarity. The samples were dissolved in methanol (1 μg μl−1) and were examined utilizing the direct inlet probe technique at an infusion rate of about 0.6 ml min−1. The optimized source parameters were as follows: octopole RF amplitude 200.0 Vpp; lens 2−60.0 V; capillary exit 154.2 V; skimmer 40.0 V; lens 1−5.0 V; Oct 1 DC 12.00 V; Oct 2 DC 2.34 V; Dry temp (set) 350 °C; Nebulizer (set) 40.0 Psi; Dry gas (set) 9.0 l min−1; HV capillary −3500 V; and HV end plate offset −500 V. Data acquisition and analysis were accomplished with Bruker Daltonics Data Analysis 3.3 software (Bruker Italia, Milano, Italy). LC-MS analyses: A symmetry C18 (Waters) column (150 mmx2.1 mm i.d., 3.5 μm) was employed; 0.1% acetic acid aqueous phase:acetonitrile:tetrahydrofuran (58:15:27 in volume) was used as mobile phase at a flow rate of 0.3 ml min−1 at 50 °C. A total of 10 μl of a sample solution in acetonitrile:water 1:1 v/v (1 mg ml−1) was injected. Both MS and diode array detectors were employed.
NMR spectroscopy
1H NMR analysis was performed at 500 MHz with a Bruker FT-NMR AVANCETM DRX500 spectrometer using a 5 mm z-PFG (pulsed field gradient) broadband reverse probe at 26 °C. The signals were unambiguously assigned by 2D COSY and HSQC experiments (the standard BRUKER pulse program). Chemical shifts are reported as δ (p.p.m.) relative to residual pyridine fixed at 7.19 (higher field signal) for 1H NMR spectra.
FT-IR spectroscopy
The infrared spectra were registered on a PerkinElmer instrument (mod. FT-IR spectrum one, PerkinElmer, Waltham, MA, USA) equipped with universal attenuated total reflection (ATR) sampling.
Optical rotation measurements
The values of optical rotation were registered on a PerkinElmer Instrument (mod. 343) at 25 °C setting the wavelength at 589 nm.
Melting-point measurements and differential scanning calorimetry
The Differential Scanning Calorimetry was registered on a PerkinElmer instrument (mod. DSC 7) at a heating rate of 10 °C min−1 from 50 to 300 °C.
The uncorrected melting points were determined by means of a Buchi B-540 instrument (Buchi, Flawil, Switzerland) capillary method, with heating rate of 5 °C min−1.
Experimental procedure
23,24-anhydro tacrolimus (5)
To a solution of tacrolimus 1 (5 g, 6.2 mmol) in toluene (100 ml), p-toluensulfonic acid monohydrate (0.255 g, 0.2 eq) was added. The solution was kept under stirring at reflux (2 h) monitoring the reaction progress by TLC (hexane:acetone 2:1). An additional amount of p-toluensulfonic acid monohydrate (0.255 g) was added; after another 2 h, when the disappearance of the starting material was observed, the reaction mixture was cooled at room temperature and purified by silica gel column chromatography. Elution with hexane:acetone (8:2) afforded a white foam that was twice crystallized (acetone:hexane 1:1). Pure 23,24-anhydro tacrolimus was recovered (1.3 g, 27%).
39,40-dihydro tacrolimus (7)
To a solution of tacrolimus 1 (3 g, 3.73 mmol) in ethyl acetate (120 ml), 10% palladium on charcoal (Fluka, St Gallen, Switzerland) was added (50% water, 0.6 g). The mixture was hydrogenated at atmospheric pressure and room temperature (2 h). The catalyst was removed by filtration through a celite pad that was washed with ethyl acetate (2 × 20 ml). Evaporation of the solvent at reduced pressure afforded pure title compound (2.8 g, 93%). TLC dichloromethane:methanol 95:5.
Purification of tacrolimus (1) by silver nitrate-impregnated silica gel column chromatography
Silver nitrate (5 g) was dissolved in acetonitrile (50 ml) in a flask protected from direct light exposure. The solution was added to silica gel (100 g, 70–230 mesh, Merck) in a round-bottom flask protected from direct light exposure. The mixture was kept under stirring until homogeneous. The solvent was removed at reduced pressure and the solid was poured in a column chromatography (stationary phase 44 × 2.9 cm2). Crude tacrolimus was dissolved in ethyl acetate:hexane 7:3 v/v to obtain a final concentration of 25% w/v and then was loaded into the column (relative ratio between crude tacrolimus:silica gel 1:37 w/w). Isocratic elution: ethyl acetate:hexane 7:3 v/v. The volume of each fraction corresponded to half of the stationary phase volume. The eluted fractions were preliminarly analyzed by TLC (eluant ethyl acetate:methanol 95:5 v/v) and the selected fractions containing tacrolimus by HPLC (‘in-house’ method).
References
Fung, J. J. Tacrolimus and transplantation: a decade in review. Transplantation 77, S41–S43 (2004).
Namiki, Y., Kihara, N., Koda, S., Hane, K. & Yasuda, T. Tautomeric phenomenon of a novel potent immunosuppressant (FK 506) in solution. J. Antibiot. 46, 1149–1155 (1993).
Tanaka, H. et al. Structure of FK 506: a novel immunosuppressant isolated from Streptomyces. J. Am. Chem. Soc. 109, 5031–5033 (1987).
Tacrolimus in US Pharmacopoeial Forum. In-process Revision. 35, 310–311 (2009).
Tacrolimus in US Pharmacopoeial Forum. In-process Revision. 36, 1566–1570 (2010).
Ham, Y-B. & Koo, Y-M. Identification of an 18-methyl derivative of tacrolimus in Streptomyces clavuligerus CDK-1119. Bull. Korean Chem. Soc. 32, 109–112 (2011).
Ferraboschi, P., Colombo, D., De Mieri, M. & Grisenti, P. First chemoenzymatic synthesis of immunomodulatingmacrolactampimecrolimus. Tetrahedron Lett. 50, 4384–4388 (2009).
Wiedeman, P. E. et al. Retention of immunosuppressant activity in an ascomycin analogue lacking a hydrogen-bonding interaction with FKP12. J. Med. Chem. 42, 4456–4461 (1999).
Akashi, T., Nefuji, T., Yoshida, M. & Hosoda, J. Quantitative determination of tautomeric FK506 by reversed-phase liquid chromatography. J. Pharm. Biomed. Anal. 14, 339–346 (1996).
Nishihara, Y. et al. (Fujisawa Pharmaceutical Co, Ltd.). Method for separating analogous organic compounds.W.O. 71,546, November 30 (2000).
Wen, Y. et al. Application of silver nitrate in the separation of macrolide antibiotic components by high-speed counter-current chromatography. J. Chromatogr. A 1216, 4668–4672 (2009).
Nakatsuka, M. et al. Total synthesis of FK506 and a FKBP probe reagent, (C8, C9-13C2)-FK506. J. Am. Chem. Soc. 112, 5583–5601 (1990).
Or, Y.S. et al. The chemistry of ascomycin: structure determination and synthesis of pyrazole analogues. Tetrahedron 49, 8771–8786 (1993).
Arai, T., Koyama, Y., Suenaga, T. & Honda, H. Ascomycin, an antifungal antibiotic. J. Antibiot., Ser. A 15, 231–232 (1962).
Okuhara, M., Tanaka, H., Goto, T., Kino, T. & Hatanaka, H. (Fujisawa Pharmaceutical Co, Ltd.). Tricyclo compounds and a pharmaceutical composition containing them. EP 184,162, June 11 (1986).
Byrne, K. M., Kaplan, L., Arison, B. H. & Colwell, Jr. L. F. (Merck & CO., INC.) Manufacture of an immunomycin homolog with Streptomyces. EP 485,091, May 5 (1992).
Acknowledgements
We thank Dr Fiorella Galimberti and Riccardo Monti for their technical assistance at the early stage of the work and Professor Ronchetti for helpful discussion.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article
Cite this article
Ferraboschi, P., Colombo, D., De Mieri, M. et al. Evaluation, synthesis and characterization of tacrolimus impurities. J Antibiot 65, 349–354 (2012). https://doi.org/10.1038/ja.2012.28
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2012.28
