
Abstract
Reproductive isolation between lineages is expected to accumulate with divergence time, but the time taken to speciate may strongly vary between different groups of organisms. In anuran amphibians, laboratory crosses can still produce viable hybrid offspring >20 My after separation, but the speed of speciation in closely related anuran lineages under natural conditions is poorly studied. Palearctic green toads (Bufo viridis subgroup) offer an excellent system to address this question, comprising several lineages that arose at different times and form secondary contact zones. Using mitochondrial and nuclear markers, we previously demonstrated that in Sicily, B. siculus and B. balearicus developed advanced reproductive isolation after Plio-Pleistocene divergence (2.6 My, 3.3–1.9), with limited historic mtDNA introgression, scarce nuclear admixture, but low, if any, current gene flow. Here, we study genetic interactions between younger lineages of early Pleistocene divergence (1.9 My, 2.5–1.3) in northeastern Italy (B. balearicus, B. viridis). We find significantly more, asymmetric nuclear and wider, differential mtDNA introgression. The population structure seems to be molded by geographic distance and barriers (rivers), much more than by intrinsic genomic incompatibilities. These differences of hybridization between zones may be partly explained by differences in the duration of previous isolation. Scattered research on other anurans suggests that wide hybrid zones with strong introgression may develop when secondary contacts occur <2 My after divergence, whereas narrower zones with restricted gene flow form when divergence exceeds 3 My. Our study strengthens support for this rule of thumb by comparing lineages with different divergence times within the same radiation.
Similar content being viewed by others
Introduction
A central assumption in evolutionary biology is that reproductive isolation accumulates with increasing genetic distance, ‘more likely as a series of small steps than in a single genetic revolution’ (Barton and Charlesworth, 1984), and thus will correlate with divergence time. However, comparative research in natural systems often neglects the time taken to speciate. In fact, diverging evolutionary lineages can be observed anywhere in the continuum from near-panmixia to various levels of increasing genetic isolation and distance, up to complete reproductive isolation. The evolutionary processes combine subtle to complex genomic changes through intrinsic mutation and drift (non-adaptive, ‘neutral’), and/or selection caused by extrinsic pressures (adaptive, ‘selective’; for example, Pereira and Wake, 2009), with ecology playing a role in most (Sobel et al., 2010).
Hybridization has complex effects on the speciation processes as recently reviewed by Abbott et al. (2013). One fascinating aspect for evolutionary research is the formation of secondary hybrid zones, in which ‘it is uncertain if barriers to gene flow will be strengthened or broken down due to recombination and gene flow’ (Abbott et al., 2013). While theory and empirical evidence suggest the latter is more likely, strongly selected genomic regions might pose exceptions (Abbott et al., 2013). Despite the potential stochastic occurrence of single large-effect factors (cf. Barton and Charlesworth, 1984), generally a gradual increase in reproductive isolation can be expected over evolutionary time in allopatry. When such lineages come early into secondary contact, with few if any ‘barrier loci’ (Abbott et al., 2013) evolved, gene flow may negate any incipient speciation (for example, Seehausen et al., 2008). In more advanced stages of allopatric speciation, gradual build up of many isolating factors of small effect (for example, Barton and Charlesworth, 1984 and Abbott et al., 2013) and necessary associations among such loci (Smadja and Butlin, 2011) can further contribute to reduce gene flow between diverging gene pools (Abbott et al., 2013), so that the involved genomes become less permeable and no longer merge.
Considering the likely gradual build up of reproductive isolation (for example, up to a certain ‘threshold above which observed differentiation is significantly greater than expected by neutral evolution alone’, Nosil and Feder, 2012), we suggest that in closely related lineages with known divergence times (estimated from paleogeographic scenarios or molecularly dated), the degree of natural hybridization at secondary contacts may serve to better understand the timing of onset and progression of speciation. Several amphibians, particularly anurans, offer suitable species systems to test this assumption, but studies on incipient speciation in this group have rarely been performed.
Meta-analyses of anuran (bufonid) breeding experiments suggest that reproductive isolation (measured by reduction in hatching success, number of larvae produced, and percentage of metamorphosis) increases with phylogenetic distance (Malone and Fontenot, 2008). It appears that very large time spans (>8 My) are required to achieve hybrid infertility or inviability. We recently discussed such laboratory data on anurans (Colliard et al., 2010), with divergence time estimates from immunological (Wilson et al., 1974), allozyme (Sasa et al., 1998) and mitochondrial distances (Malone and Fontenot, 2008), or mitochondrial and nuclear sequence data (Sumida et al., 2007), showing that in the laboratory some anurans may still produce viable F1 offspring after >20 My divergence, but may develop ‘partial or complete hybrid inviability’ after >8 My divergence (Sumida et al., 2007). However, under natural conditions, several empirical studies on anurans suggest reproductive isolation to have arisen after divergence initiated in the Pliocene (5.3–2.6 My) or earlier, as best studied in parapatric Bombina (for example, Szymura, 1993, Kruuk et al., 1999, Vines et al., 2003 and Hofman et al., 2007), assumed to have diverged between Upper Miocene and Lower Pliocene (B. bombina–B. variegata, 8.96 (12.74–4.93) My or 6.48 (8.89–4.19) My, depending on two different calibration settings; Pabijan et al., 2013). Similar time frames were estimated for single hybrid systems of Australian and European hylid frogs (Hoskin et al., 2005; Verardi et al., 2009), South-American dendrobatids (Simões et al., 2012) and green toads (Colliard et al., 2010). By contrast, case studies on clades of more recent, Pleistocene (2.5 My–11 Ky) divergence (for example, 1.33 My in ranid frogs, Canestrelli and Nascetti, 2008; 1.69–0.33 My in bufonid toads, Sequeira et al., 2011) may form ‘wide hybrid zone(s) with a considerable genetic exchange between both gene pools’ (Santucci et al., 1996), where reproductive barriers could be weak or absent.
One challenge for testing an inverse relationship between divergence time and natural hybridization in anuran amphibians in the long term is the disconnected evidence from multiple (often) distantly related species groups. The radiation of Palearctic green toads (Bufo viridis subgroup) offers an excellent opportunity as it includes several lineages that arose at different times and form secondary contact zones. Specifically, the central and northeastern Mediterranean shores, the Apennine Peninsula and Sicily, are inhabited by four lineages with secondary contacts bringing together pairs with three different Plio-Pleistocene divergence times (Stöck et al., 2006, 2008a, 2008b). These are Bufo variabilis (PALLAS, 1769) on the eastern Balkan Peninsula, in Asia Minor, and northern Central Asia, B. viridis (LAURENTI 1768), on the western Balkan Peninsula and in Central Europe, B. balearicus (BOETTGER, 1880) on the Apennine Peninsula, Corsica, Sardinia and the Balearic Islands, and finally B. siculus STöCK, SICILIA, BELFIORE, BUCKLEY, LO BRUTTO, LO VALVO, ARCULEO 2008, in Sicily.
We have recently shown that the Sicilian endemic B. siculus and the Italian mainland-origin B. balearicus, with an estimated divergence time of 2.7 My (4.9–1.1, Stöck et al., 2008a), form a narrow hybrid zone east of Mt. Etna. Analysis of nuclear genomes showed very sharp transition at the contact, with essentially no admixture (Colliard et al., 2010). The highest pairwise FST values (>0.50) were found between populations from each side of this contact zone (only ∼16 km apart). All individuals from these populations were assigned with maximum likelihood (100%) to either B. siculus or B. balearicus, respectively. MtDNA analyses evidenced some limited bidirectional introgression over∼40 km, with few cases of cyto-nuclear discordances, testifying to rare events of past hybridization, but no wild-caught F1 individuals. Altogether, this analysis suggested very low, if any, current gene flow, as furthermore supported by several experimental crosses, showing strong hybrid breakdown in F2 and backcrosses (Colliard et al., 2010).
In the present paper, we compare signatures of secondary contact and introgression in Sicily with another contact zone in northeastern Italy, where the same lineage B. balearicus meets another but more recently diverged lineage, namely B. viridis. As we show, this contact zone offers a striking contrast to the Sicily situation, with much wider introgression both at nuclear and at mitochondrial levels, suggesting that gene flow is barely constrained by intrinsic genomic incompatibilities in these less differentiated lineages.
Materials and methods
Sampling
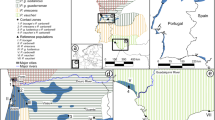
Samples (buccal swabs from adults and subadults; muscle from road kills and scientific vouchers; tail tips from tadpoles) were collected during fieldwork (2008–2010; Figure 1, Table 1; Supplementary Table S1), or came from scientific collections (Museo di Storia Naturale di Venezia, MSNVE; Museo Civico di Storia Naturale di Ferrara, MCSNFE). Overall, 316 specimens from 63 localities were available across the eastern Po Plain, between the northern Apennine and the Venetian Pre-Alps. The majority of samples per population (term used in the sense of ‘locality sample’ throughout this paper) came from single sampling sites. As long as no potential barriers like rivers (see Discussion) or highways (for example, Forman et al., 2003) were in between, very few samples from neighboring sites were pooled (Supplementary Table S1) if their distance was <2 km, corresponding to the lower limit of the migration distance of green toads in a single year (2–10 km; Blab et al., 1991). This seems justified because the Po Plain is otherwise almost free of elevation differences and major natural barriers for amphibian movement. For tadpoles, to avoid the collection of siblings, samples were taken from unconnected ponds or, in rare instances, where it was not possible, very distant positions within the same pond, and in such cases always from differently sized cohorts.

Assignment to Bufo balearicus or B. viridis for all populations, based on Bayesian clustering (STRUCTURE) of 12 microsatellite genotypes. Assignment per population (pie charts) corresponds to the average assignment probabilities of individuals at this location to each of the two groups. Pie size proportional to sample size; bar plots from STRUCTURE to the right (K=2); approximate Bronze Age paleohydrographic system (Piovan et al., 2010) in purple.
DNA extraction and generation of genotype and sequence data
DNA was extracted using the Qiagen DNeasy kit. Twelve microsatellites, polymorphic in both species, were amplified for 254 samples as described (Colliard et al., 2010, Dufresnes et al., 2011). In 287 individuals, we sequenced ca. 860 bp of the mitochondrial D-loop, according to Stöck et al. (2006). For direct comparisons, only a fragment (591 bp) of this, also available from a previous study (Stöck et al., 2008a), could be included to estimate the divergence time. We also cloned and sequenced 580 bp of a sufficiently variable nuclear intron of alpha-Tropomyosin (details: Stöck et al., 2008a), in 13 individuals from the contact zone and representatives of both species from throughout their distribution ranges.
Sequence alignment and phylogenetic analyses
Sequences were edited in SEQUENCHER v. 4.9 (Gene Codes) and aligned using SEAVIEW v.4.2.4 (Gouy et al., 2010). For the population analyses of mtDNA, we used the program TCS v.1.21 (Clement et al., 2000). Phylogenetic analyses were made using PHYML, v.2.4.5 as implemented in SEAVIEW (Gouy et al., 2010), and HKY+G models (JMODELTEST v.0.1.1; Posada, 2008) for both mtDNA and nuDNA. We choose a BioNJ as a starting tree, and used the combined subtree pruning and regrafting plus nearest neighbor interchange options for tree improvement; otherwise, default parameters were used (http://atgc.lirmm.fr/phyml/ for details). We generated bootstrap values based on 1000 resampled data sets.
Molecular dating
Molecular dating for major mitochondrial and nuclear lineages was performed using BEAST v.1.6.1 (Drummond et al., 2007; http://beast.bio.ed.ac.uk/). To obtain relative divergence time estimates for the most recent common ancestor of B. viridis vs B. balearicus, we calibrated the previously obtained divergence date for B. siculus vs B. boulengeri at 1.8 My (3.5–0.63, 95% highest posterior density intervals, HPDIs; Stöck et al., 2008a), and the divergence of lineages boulengeri-siculus vs balearicus-viridis at 2.75 My (4.9–1.18) on a previously used fragment of the mtDNA control region (591 bp; Stöck et al., 2008a), assuming an uncorrelated lognormal relaxed molecular clock and a Yule birthrate tree prior (constant speciation rate per lineage) as most appropriate for species level divergences (Drummond et al., 2007). We applied the same calibrations for the nuclear data set (alpha-Tropomyosin). We used an UPGMA starting tree. Two independent analyses for 50 million generations were run with tree sampling every 1000 generations. Convergence and stationarity were checked in the program TRACER v.1.5. Results were combined in the BEAST module LOGCOMBINER v.1.6.0. The ‘burn-in’ value was selected after visualizing log likelihoods associated with the posterior distributions of trees in TRACER. All trees generated before the log likelihood curve flattened out were discarded.
Demographic analyses
To calculate the distributions of observed and expected pairwise nucleotide site differences between individual mtDNA haplotypes under a model of demographic expansion (mismatch distributions), we used DNASP v.5 (Librado and Rozas, 2009). We included only D-loop markers for which 874 bp 100% readable sequences were available. Ages of expansions were calculated from the parameter τ, estimated by DNASP (τ=2 μt, where μ is the substitution rate and t is the time since expansion), using a substitution rate for the D-loop of ca. 2% per million years (Stöck et al., 2008a). In addition, we computed (DNASP) the following tests of selective neutrality: Fu’s Fs, Tajima’s D and Ramos-Onsins & Rozas’s R2 (Ramos-Onsins and Rozas, 2002 and references therein); significances were tested by coalescent simulations (10 000 replicates).
Genotype data analyses
We used MICRO-CHECKER v.2.2.3 (Van Oosterhout et al., 2004) to exclude genotyping errors due to null alleles, stuttering and allelic dropout. Using FSTAT v.2.9.3 (Goudet, 1995), we tested for linkage disequilibrium between each pair of loci in each population. Hardy–Weinberg equilibrium and pairwise differentiation (FST) were assessed for populations with sufficient sample sizes (n⩾5) in ARLEQUIN v.3.5 (Excoffier et al., 2005). For these populations, we also computed allelic richness (A. r.) and observed heterozygosity (He) with FSTAT, which performs a rarefication procedure to a common sample per locus. Analyses of microsatellite genotypes provided no evidence for large allelic dropout from any locus or population.
We used the Bayesian clustering algorithm STRUCTURE v.2.3 (Pritchard et al., 2000) to assess interspecific and intraspecific genetic structures of populations based on microsatellite genotypes. We applied the admixture model and allowed for correlated allele frequencies between populations, as recommended for cases of subtle population structure. A range of different cluster sizes (K) from 1 to the number of localities per analysis was tested. Each run was replicated 10 times with 105 iterations following a ‘burn-in’ period of 104. To infer the number of clusters (K) that best fitted our data, we applied the ΔK ad hoc statistics (Evanno et al., 2005).
In interspecific analyses, individuals were considered as hybrids if their STRUCTURE assignment probability to either cluster (K=2) was <0.9, with 90% credible intervals (CIs) neither reaching 0 nor 1, or if they were assigned by STRUCTURE to one clade (that is, nuclearly ‘pure’: 90% CI within 0.9 and 1) but contained the mitochondrial haplotype of the other lineage (cyto-nuclear discordance). Considering CIs allows distinguishing between individuals that harbor alleles uninformative about the source taxa, and confidently assigned hybrid or ‘pure’ individuals (for example, Sá-Pinto et al., 2010). To get insights into the nature of hybrids, we analyzed our microsatellite data set with NEWHYBRIDS v.1.1 (Anderson and Thompson, 2002), which computes the Bayesian posterior probability of assignment of each individual to several genotypic classes (parental, F1, F2, backcrosses). Runs were repeated with various numbers of iterations. Several individuals were pre-assigned as ‘parents’ (using the z parameter), but we restricted this pre-assignment to individuals whose assignment by STRUCTURE was higher than 0.99 (with 90% confidence interval within 0.9 and 1), to discard potential highly backcrossed individuals. We also conducted a principal component analysis using PCAGEN v.2 (Goudet, 1999) on allelic frequencies to visualize population differentiation. Significance of axes was tested by 10 000 randomizations of genotypes.
Geographic transects
We conducted additional STRUCTURE analyses along three transects evenly distributed in the flat corridor delimited by the Pre-Alps in the northwest and Adriatic Sea in the east, crossing the Po and Adige rivers (Figure 2; A−C), and presumably corresponding to major, topographically possible migration directions of the two toad lineages (transect A: locs. 1–4, 17, 25–26, 28, 47–52, 56; transect B: locs. 1–4, 18, 24, 27, 29–33, 43–46, 53–55; transect C: 5, 6, 8–15, 23, 34–40, 57–60). Along transects, the frequencies of alleles diagnostic for B. balearicus (that is, absent from pure viridis populations) were calculated for localities with n⩾4 (transect A: locs. 1–4, 17, 26, 28, 47, 50, 56; transect B: locs. 1–4, 30, 31, 43, 53–55; transect C: 5, 6, 8–15, 23, 34–40, 57–60). For two transects (A, B; C was dismissed due to a sampling gap in its southern part), we computed genetic clines for the mitochondrial marker and for microsatellites possessing species-specific alleles (that is, alleles that contributed to cluster differences and for which frequencies, when narrowed down to a two-allele system, were higher than 0.9 in pure populations of one species). We further cross-checked the diagnostic value of alleles according to coordinates on the first axis of a multiple correspondence analysis (GENETIX v.4.05; Belkhir et al., 1998) of allelic frequencies of pure populations (Gay et al., 2008). For each microsatellite, selected alleles were assigned to species-specific compound alleles to reduce the variation to a two-allele system. Along each transect, clines were fitted to allelic (microsatellites) and haplotype (mtDNA) frequencies with the program CFIT v.7.0 (Gay et al., 2008), using a three-part stepped cline model, comprising a central sigmoid and two exponential tails (Szymura and Barton, 1986). We performed a likelihood search for a common center (coincidence) and slope (concordance) of all clines (Gay et al., 2008), reiterating each fit with different random seeds to check for convergence. Models with different constraints (common center, common slope and both) were successively fitted to all markers simultaneously, and likelihood-ratio tests were performed to compare constrained with unconstrained clines. In the final models, individual clines, for which the constrained models were rejected (likelihood-ratio tests significant, 5% level), were fitted independently. For each transect, we chose the final model with the lowest Akaike information criterion (AICc).

Frequency of mitochondrial D-loop haplotypes for all populations (map), mismatch distributions of three mtDNA haplotype groups (corners), and microsatellite genotypes (STRUCTURE assignment probabilities) along three transects A, B and C (barplots). Pie size is proportional to mtDNA sample size; approximate Bronze Age paleohydrographic system (rivers, coast line; Piovan et al., 2010) in purple; relative locations of the modern Po and Adige Rivers in blue (in purple for paleo-rivers) within bar plots; mismatch distributions: the dotted line shows the frequency distribution of the observed pairwise differences; the solid line shows the frequency distribution of the expected pairwise differences under the sudden expansion model.
To examine isolation by distance and the role of major rivers (Adige, Po) as potential barriers to dispersal, we computed partial correlations between pairwise FST, geographic distances and a matrix of the number of rivers between populations (n⩾5) by partial Mantel tests (ARLEQUIN). The geographic distance matrix was obtained using GEOGRAPHIC DISTANCE MATRIX GENERATOR v.1.2.3 (Ersts, 2006). Significance of correlations was tested by a permutation procedure (10 000 permutations).
Results
Phylogenetic analyses and divergence times
The viridis mtDNA clade is homogenous across the study region, and reveals a significant range expansion that occurred ca. 16 Ky ago (Figure 2; Table 2). In contrast, the balearicus haplotypes form three subclades (1a, 1b and 2; Figure 2; Supplementary Figure S1) with some geographic structure. Two subclades show significant signs of expansions (Figure 2; Table 2), dating back to 43 Ky ago for haplotype 1A (the most northern), but only 3 Ky for haplotype 1B (Figure 2). Transects show a large region of co-occurrence and a smooth transition in frequencies over >100 km from balearicus to viridis haplotypes. Networks for the major clades are provided with maximum-likelihood bootstrap support in Supplementary Figure S1 (see also trees in Stöck et al., 2006, 2008a).
Nuclear sequences (alpha-Tropomyosin intron) also differentiate two highly supported clades, corresponding to B. viridis and B. balearicus, respectively (Figure 3). The B. balearicus clade is widespread on the Apennine Peninsula, from the Po Plain (study area) to southern Italy, and also found on Sardinia, the Balearic Islands and easternmost Sicily. Interestingly, two individuals (Po16, loc. 30; BV247, loc. 1, Figure 3) harbored an allele from balearicus as well as viridis, demonstrating their nuclear hybridity.

Phylogenetic tree based on a nuclear sequence marker. Maximum likelihood tree based on clones (‘cl’) obtained from 580 bp of an intron of alpha-Tropomyosin. Sample number (sometimes several with same haplotype and locality) is followed by locality information and population number (as in Figure 1, Supplementary Table S1); individual ‘Po2’, highlighted green, possessed a D-loop of one lineage but Tropomyosin alleles from the opposite species; toads BV247 and Po16 each contained an allele from balearicus and viridis, respectively.
Molecular dating of mitochondrial and nuclear sequence data using BEAST suggests a Lower Pleistocene divergence of B. viridis and B. balearicus. Estimates point to 1.9 My (95% HPDI: 2.5–1.3 My) for the mtDNA D-loop, and 2.0 My (95% HPDI 3.04–1.09 My) for intronic sequences of the nuclear Tropomyosin. The posterior predictions for the divergence time between B. siculus and B. balearicus were very close to the mode assumed for the prior, and consistent between mtDNA and nuDNA: namely 2.65 (95% HPDI: 3.3–1.9 My) and 2.5 My (95% HPDI: 3.5–1.55 My) for the D-loop and the Tropomyosin, respectively.
Population structure
In two populations, assigned to B. balearicus (pop. 50, 56), null alleles were detected for locus C201 and corrections performed. We did not find significant linkage disequilibrium in any population after sequential Bonferroni corrections. All but one population (pop. 40, B. balearicus) met Hardy–Weinberg expectations. Analyses of microsatellite genotypes using STRUCTURE clearly grouped individuals into two clusters (K=2, best fitting the whole data set), corresponding to viridis and balearicus gene pools (Figure 1). Most individuals from the northeastern part of the study area (north of Euganei hills and Venice; pop. 1–4, 7–8, 12–13, 15) were assigned to pure viridis, whereas individuals from south of the Po River were mainly assigned to pure balearicus (pop. 50, 52, 54–56, 58). Individuals from localities in between showed intermediate but robust probabilities of assignment (that is, 90% CI neither reaching 0 nor 1), suggesting clear signs of nuclear admixture (Figure 1).
Pairwise FST values revealed a clear pattern of isolation by distance, as illustrated by the principal component analysis (Figure 4). The first and only significant axis explaining 33.6% of the variance (P=0.0001, bootstrapping) opposes pure viridis (right) to pure balearicus (left) populations. The transition in-between is continuous through the contact zone. The populations farthest away from the contact zone center display the highest FST values (0.53), whereas those at the contact zone show little or no differentiation (FST 0.00–0.03; Supplementary Table S2).

Principal component analysis on allelic frequencies including all populations, using PCAGEN (Goudet, 1999). Only the first axis is significant (P<0.01); green/red coloration is proportional to average assignment probability to B. viridis/B. balearicus, respectively (see Figure 1).
Isolation by distance was confirmed by Mantel tests; over the whole data set, pairwise FST increased significantly with geographic distance (44% of the variance explained, P<0.001). In addition, partial Mantel tests revealed a significant effect of major rivers (Po and Adige). Including the present-day riverbeds explained 61% of the variance, of which 25% was due to geographic distance, and the remaining (36%) to the number of large rivers (0, 1 or 2) separating populations. Interestingly, very close and significant values are also obtained when historical (Bronze Age), rather than present-day riverbeds are included (Table 3).
Genetic diversity was quite uniform over the study region, although values at the contact zone (He ⩾0.60) were slightly higher than in the pure viridis or balearicus range (Table 1).
Cline analyses
Seven microsatellites with at least one species-specific allele (STRUCTURE) and the mitochondrial marker were selected for cline analyses (Figure 5; Supplementary Table S3). The overall pattern shows two extrinsic effects: that of geographic distances and that of riverbeds (Figure 5). The proportion of balearicus-specific alleles decreases smoothly with increasing latitude, with marked drops corresponding approximately to the (historic beds of) Po and Adige Rivers. No viridis-specific nuclear alleles were detected south of the Po River. At both transects, the best models had a shared center and a shared slope for six out of the seven nuclear markers (Supplementary Table S4, AICc). The common center was located between the two major rivers (20.0 km northeast of the Po for transect A and 20.5 km for transect B; that is, near the historic Po River bed). Cline widths, calculated as the inverse of the slope, were remarkably narrow (2.3 km for transect A, 2.6 km for transect B; Supplementary Table S3), given that B. viridis can migrate 2–10 km in a single year (Blab et al., 1991), but flanked with large introgression tails, expanding northwards of Adige into the viridis range (Figure 5). For the mtDNA, the shape of the clines differed strongly from those of nuclear markers, with centers of mtDNA clines located much further north (46.6 km northeast of the Po for transect A; 96.4 km for transect B).

Genetic clines for seven microsatellite loci with species-specific alleles and the mitochondrial control region (D-loop) along transects A (a) and B (b). Relative position of the Po and Adige rivers (current and, if different within the transect, paleo-river beds) is plotted as dashed lines. Transects are the same as in Figure 2, but including only localities where n⩾4.
Additional evidence for wide asymmetric introgression and backcrosses
Many cases of cyto-nuclear discordances were detected, with an asymmetric distribution. Of the individuals confidently assigned to viridis by nuclear markers (51 inds.), 29% possessed balearicus mtDNA (15 inds.), including some from populations distant from the center of the contact (pops. 3–4, 7–8; Figure 1). However, none of those confidently assigned to balearicus (23 inds.) presented viridis mtDNA (all possessed balearicus mtDNA). NEWHYBRIDS also identified many hybrids at localities displaying admixture: populations 22–35 had 42% of F2 hybrids and 16% of backcrosses, while populations 37–48 had 59% F2 hybrids and 3–5% of backcrosses (Supplementary Figure S2). No F1 hybrids were detected. The geographic distribution of the Tropomyosin alleles also provides evidence for introgression, with admixed populations mostly at the center of the contact zone.
Discussion
Differences in the degree of introgression
As our results show, the Italian (B. balearicus) and the European green toad (B. viridis) come into parapatry in the lower Po and Adige drainages. Despite substantial mitochondrial and nuclear sequence differentiation, accumulated over 1.9 My divergence (Figure 3 and Supplementary Figure S1; see also Stöck et al., 2008a), these two lineages show extensive hybridization in their secondary contact zone. This situation contrasts strikingly with the one documented in Sicily between B. balearicus and B. siculus, two lineages with a deeper divergence (2.6 My). MtDNA introgression was limited to 40 km in Sicily (Colliard et al., 2010), but stretches up to 130 km in the Po Plain (Figure 2). Similarly, nuclear admixture (that is, >10% assignment probability to the alternative taxon) was almost absent in Sicily, but expands over 40–50 km in the balearicus/viridis contact zone (Figure 1).
This contrast is reflected in the patterns of isolation by distance: in Sicily, pairwise FST reach their highest values (0.50) between the siculus and balearicus populations immediately adjacent to the contact zone (only ∼16 km apart), but increase smoothly with geographic distance in the balearicus/viridis contact zone, reaching their maximum (FST=0.53) between the most distant populations. Genetic diversity shows a similar contrast: in Sicily, He values were the lowest at the balearicus/siculus contact zone (Colliard et al., 2010), but were largely uniform over the Po Plain, with slightly higher values (He⩾0.60) at the viridis/balearicus contact zone. This and the advanced degree of hybridization in the Po Plain zone, including many individuals exhibiting a cyto-nuclear discordance, suggest that no major pre- and post-zygotic reproductive barriers limit the gene flow between both species, although detailed bioacoustic studies and examination of Haldane effects (see below) are still missing. Taken together, all population genetics parameters reveal two clearly dissimilar situations: introgression under secondary contacts is still extensive between B. balearicus and B. viridis (1.9 (2.5–1.3) My divergence) but virtually absent between B. balearicus and B. siculus (2.6 (3.3–1.9) My divergence).
Although our results are well in line with the relatively few scattered studies in anurans in which the relationship between divergence time and natural degree of hybridization has been studied (see Introduction), the two hybrid zones compared so far do not allow us to draw general conclusions yet. Nevertheless, we note that narrow hybrid zones form in secondary contacts of lineages with Pliocene divergence (>3 My; Szymura, 1993; Hofman et al., 2007, Verardi et al., 2009, Simões et al., 2012), whereas secondary contacts of lineages with more recent Plio-Pleistocene divergence (2.5 My–11 Kya) result in wide(r) hybrid zones with considerable genetic exchange between both gene pools (Santucci et al., 1996, Canestrelli and Nascetti, 2008; Sequeira et al., 2011). While few if any comparative studies within the same radiation have been undertaken in anurans, among urodeles, the Ensatina ring species complex presents a famous system, in which ‘extant intermediate stages of terminal forms have a nearly continuous range, offering replicated interactions at several stages of divergence’ (Pereira and Wake, 2009). Reproductive isolation in Ensatina ‘is likely to be a byproduct of processes that contribute to overall [nuclear] genetic divergence, such as time in geographic isolation’ (Pereira et al., 2011), and recent evidence supports ‘asymmetric reproductive isolation between terminal forms’ of the ring (Devitt et al., 2011).
Beyond divergence time, additional specificities of the two green toad hybrid zones may contribute to the observed introgression differences. Island populations, like that of B. siculus, can experience increased drift and selection and thus accelerated rates of molecular evolution (for example, Woolfit and Bromham, 2005), which could enhance the incompatibility between B. balearicus and B. siculus gene pools. Moreover, we assume that the time since toad lineages came first into secondary contact in Sicily dates at least back to low sea levels during the Last Glacial Maximum (20 Kya; allowing B. balearicus to colonize Sicily). Signatures of population expansion (43–3 Kya; see above; and river translocations, see below) point to similar, and thus an overlapping time period for the first contact between B. balearicus and B. viridis in the Po Plain. Therefore, the age of contact zones would hardly explain the contrasting introgression patterns.
Hybrid zone movement, geography and colonization history
Gene flow in the Po Plain seems barely restricted by genomic incompatibilities and rather reflects the history of colonization and dispersal effects as well as fine scale influences of geographic features.
Interestingly, the clines of nuclear markers are asymmetric, with a long tail on the viridis side (Figure 3). A tail of clines of unlinked neutral markers with apparent unidirectional introgression across the zone (Moran, 1981, cited in Buggs, 2007) is typical of a ‘moving hybrid zone’. Barton and Hewitt (1985) suggested that evidence for hybrid zone movement should be based on many neutral alleles, introgressing in the same direction. This is the case for balearicus microsatellite alleles and suggests zone movement as partial explanation for the introgression asymmetry and tail at the viridis side of the zone in the Po Plain. This evidence is further supported by an asymmetry of nuDNA vs mtDNA introgression, again with a tail at the viridis side of the hybrid zone. Contrasting with most nuclear markers that share a common center (20 km north of the modern Po river), the mtDNA cline centers much further north (Figure 5), in line with the patterns of cyto-nuclear discordances: we found many balearicus mtDNAs in a viridis nuclear background but not the reverse.
Beyond introgression asymmetry and isolation by distance, the balearicus/viridis hybrid zone reflects signatures of physical barriers (Figures 2 and 5) to gene flow imposed by the current and historic position of large rivers (Po and Adige). Partial Mantel-tests could assign 36% of the variance in pairwise FST to the number of main (or historic) rivers (0, 1 or 2), separating populations. Cline analyses also showed marked drops in the frequency of balearicus specific alleles, corresponding to these main, current or historic, rivers (Figure 5). Intriguingly, all hybrid populations showing nuclear introgression (Figure 1) are almost perfectly delimitated to the south by the modern Po river, and to the northeast by the ancient Po and Adige riverbeds, which, some 5 Kya, were situated 20–40 km farther northeast than today (pink in Figures 1 and 2; Piovan et al., 2010). As the Mantel tests supported, the locations of historical riverbeds were only slightly less important than modern ones in accounting for present day population structure (Table 3).
All of this suggests the following scenario for this secondary contact zone. Since the Last Glacial Maximum (LGM), the Po Plain vegetation changed from a natural steppe-like to a densely forested habitat. This was followed some 4–5 Ky ago by human deforestation (Amorosi, 2004), which presumably facilitated green toad range expansion. The region was progressively colonized by balearicus and viridis from their glacial refugia, respectively, south and east of the study area. The contact zone might have been established at the Po and/or Adige paleo-riverbeds, which at this time posed faster running river barriers than today (Fontana et al., 2008), with relatively few, but some migrants overcoming them (for example, balearicus may have expanded further north). A 20–40 km southward river translocation into the modern beds then trapped some balearicus populations northeast of the present-day Po River. Continuous gene flow from incoming northern viridis colonizers progressively diluted local balearicus genomes, now isolated by the Po River from the main range of balearicus. The long tail of mtDNA and nuDNA clines in the north might be a result of this influx of viridis genes into the hybrid zone, explicable as ‘balearicus dilution’ itself and/or by truly southwestwards migrating viridis populations. In any case, this dynamic, apparently ongoing process exhibits the clear features of a ‘moving hybrid zone’. In accordance with a scenario of ‘balearicus dilution’, NEWHYBRIDS analyses did not detect any pure balearicus or F1 genotypes north of the Po River, suggesting instead an advanced state of nuclear admixture (‘F2’ and backcrosses; Supplementary Figure S2). The absence of ‘F1’ suggests that the individuals labeled ‘F2’ might in fact also represent backcrosses. Most importantly, these data imply that at least some hybrids become adult and fertile, as they successfully reproduce. This scenario would suggest a minimal age of 5 Ky for the hybrid zone (that is, >2500 generations, assuming a 2-year generation time; Stöck et al., 2008b), as inferred from the last major Po River translocation. The further reaching mitochondrial than nuclear introgression of balearicus into a viridis background might further indicate a smaller female effective size in viridis invaders, possibly stemming from sex differences in dispersal ability. A male-biased dispersal is expected to increase male effective population size at the front of invading populations, favoring mtDNA over nuclear introgression (Petit and Excoffier, 2009). We do not know whether slight size differences between the green toad lineages involved (Stöck et al., 2008b) could skew the mating preferences as, for example, observed within B. bufo for differently sized males (Davies and Halliday, 1979). Another alternative that could explain nuDNA/mtDNA introgression asymmetry might stem from sex differences in hybrid fitness or fecundity. Haldane’s rule predicts lower hybrid fitness in the heterogametic sex. Several taxa of the B. viridis group, probably including B. balearicus, have a male heterogametic (XY) sex determination system (Stöck et al., 2013). Although Haldane’s rule might be less prevalent in taxa with homomorphic sex chromosomes, too few non-model hybrid organisms have been studied, especially in natural systems of amphibians, to exclude this scenario (Schilthuizen et al., 2011).
Conclusions
Our data show a striking contrast in the degree of hybridization between closely related green toad lineages in secondary contact. In comparison with the system of greater divergence (Sicily, 2.6 My), the North-Italian green toad hybrid zone (of lineages diverged 1.9 My) exhibits a much wider and asymmetric introgression at nuclear and mitochondrial levels. Gene flow in this apparently dynamic system seems mainly constrained by local geographic barriers (large rivers), and less by intrinsic genomic incompatibilities. All of this suggests that reproductive isolation during incipient speciation increases gradually with the time of divergence (up to a certain threshold; for example, Nosil and Feder, 2012), and might be driven by complex genomic processes rather than single speciation genes. Our study represents a contribution toward comparative studies of secondary contacts of closely related anuran lineages. It is well in line with scattered research in other anuran species, with examples among discoglossoids, bufonids, hylids and ranids, but has the advantage to compare lineages with different divergence times from the same radiation.
Data archiving
Genotype data available from the Dryad Digital Repository: doi:10.5061/dryad.85pr1. GenBank accessions for the D-loop and alpha-Tropomyosin have in part been published by Stöck et al. (2008a) and Colliard et al. (2010); new data in the present paper have accessions: KJ532478-KJ532515 (alpha-Tropomyosin) and KJ532516-KJ532802 (D-loop).
References
Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N et al. (2013). Hybridization and speciation. J Evol Biol 26: 229–246.
Amorosi A . (2004). Po plain. In: Margottini C, Vai GB, (eds) Climex Maps Italy. Litho-Paleoenvironmental Maps of Italy During the Last Two Climatic Extremes –Explanatory notes. Museo Geologico Cappellini: Bologna. pp 38.
Anderson EC, Thompson EA . (2002). A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160: 1217–1229.
Barton NH, Charlesworth B . (1984). Genetic revolutions, founder effects and speciation. Annu Rev Ecol Evol Syst 15: 133–164.
Barton NH, Hewitt GM . (1985). Analysis of hybrid zones. Annu Rev Ecol Evol Syst 16: 113–148.
Belkhir K, Borsa P, Chikhi L, Goudet J, Bonhomme F . (1998) GENETIX 3.07, WindowsTM software for population genetics. University of Montpellier II: Montpellier, France.
Blab J, Brüggemann P, Sauer H . (1991). Tierwelt in der Zivilisationslandschaft, Teil II: Raumeinbindung und Biotopnutzung bei Reptilien und Amphibien im Drachenfelser. Land. Schriftenr. Landschaftspfl. Naturschutz (Bonn-Bad Godesberg) 34: 1–94.
Buggs RJA . (2007). Empirical study of hybrid zone movement. Heredity 99: 301–312.
Canestrelli D, Nascetti G . (2008). Phylogeography of the pool frog Rana (Pelophylax) lessonae in the Italian Peninsula and Sicily: Multiple refugia, glacial expansions and nuclear-mitochondrial discordance. J Biogeogr 35: 1923–1936.
Clement M, Posada D, Crandall K . (2000). TCS: a computer program to estimate gene genealogies. Mol Ecol 9: 1657–1660.
Colliard C, Sicilia A, Turrisi GF, Arculeo M, Perrin N, Stöck M . (2010). Strong reproductive barriers in a narrow hybrid zone of West-Mediterranean green toads (Bufo viridis subgroup) with Plio-Pleistocene divergence. BMC Evol Biol 10: 232.
Davies NB, Halliday TR . (1979). Competitive mate searching in male common toads, Bufo bufo. Anim Behav 27: 1253–1267.
Devitt TJ, Baird SJE, Moritz C . (2011). Asymmetric reproductive isolation between terminal forms of the salamander ring species Ensatina eschscholtzii revealed by fine-scale genetic analysis of a hybrid zone. BMC Evol Biol 11: 245.
Dufresnes C, Betto-Colliard C, Perrin N, Stöck M . (2011). Thirteen polymorphic microsatellite markers for the European green toad Bufo viridis viridis, a declining amphibian species. Conserv Genet Resour 3: 311–313.
Drummond AJ, Ho SYW, Rawlence N, Rambaut A . (2007) A Rough Guide to BEAST 1.4. University of Auckland: Auckland, New Zealand http://beast-mcmc.googlecode.com/files/BEAST14_Manual_6July2007.pdf.
Ersts PJ . (2006) Geographic Distance Matrix Generator (version 1.2.3). American Museum of Natural History: New York, USA http://biodiversityinformatics.amnh.org/open_source/gdmg.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Laval G, Schneider S . (2005). Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bioinform 1: 47–50.
Fontana A, Mozzi P, Bondesan A . (2008). Alluvial megafans in the Venetian-Friulian Plain (north-eastern Italy): evidence of sedimentary and erosive phases during Late Pleistocene and Holocene. Quat Int 189: 71–90.
Forman RTT, Sperling D, Bissonette JA, Clevenger AP, Cutshall CD, Dale VH et al. (2003) Road Ecology. Island Press: Washington, DC.
Gay L, Crochet PA, Bell DA, Lenormand T . (2008). Comparing clines on molecular and phenotypic traits: a window on tension zone models. Evolution 62: 2789–2806.
Goudet J . (1995). FSTAT (version 1.2): A computer program to calculate F-statistics. J Hered 86: 485–486.
Goudet J . (1999) PCAGEN v. 1.2.1. University of Lausanne: Switzerland http://www2.unil.ch/popgen/softwares/pcagen.htm.
Gouy N, Guindon S, Gascuael O . (2010). SeaView version 4: a multiplatform user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27: 221–224.
Hofman S, Spolsky C, Uzzell T, Cogalniceanu D, Babik W, Szymura JM . (2007). Phylogeography of the fire-bellied toads Bombina: independent Pleistocene histories inferred from mitochondrial genomes. Mol Ecol 16: 2301–2316.
Hoskin CJ, Higgie M, McDonald KR, Moritz C . (2005). Reinforcement drives rapid allopatric speciation. Nature 437: 1353–1356.
Kruuk LEB, Gilchrist JS, Barton NH . (1999). Hybrid dysfunction in fire-bellied toads (Bombina). Evolution 53: 1611–1616.
Librado P, Rozas J . (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Malone JH, Fontenot BE . (2008). Patterns of reproductive isolation in toads. PLoS One 12: e3900.
Nosil P, Feder JL . (2012). Genomic divergence during speciation: causes and consequences. Philos Trans R Soc B 367: 332–342.
Pabijan M, Wandycz A, Hofman S, Wecek K, Piwczynski M, Szymura JM . (2013). Complete mitochondrial genomes resolve phylogenetic relationships within Bombina (Anura: Bombinatoridae). Mol Phylogenet Evol 69: 63–74.
Petit RJ, Excoffier L . (2009). Gene flow and species delimitation. Trends Ecol Evol 24: 386–393.
Pereira R, Wake DB . (2009). Genetic leakage after adaptive and non-adaptive divergence in the Ensatina eschscholtzii ring species. Evolution 63: 2288–2301.
Pereira R, Monahan WB, Wake DB . (2011). Predictors for reproductive isolation in a ring species complex following genetic and ecological divergence. BMC Evol Biol 11: 194.
Piovan S, Mozzi P, Stefani C . (2010). Bronze age paleohydrography of the southern Venetian Plain. Geoarchaeology 25: 6–35.
Posada D . (2008). jModelTest: phylogenetic model averaging. Mol Biol Evol 27: 1253–1256.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Ramos-Onsins SE, Rozas J . (2002). Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19: 2092–2100.
Santucci F, Nascetti G, Bullini L . (1996). Hybrid zones between two genetically differentiated forms of the pond frog Rana lessonae in southern Italy. J Evol Biol 9: 429–450.
Sá-Pinto A, Baird SJE, Pinho C, Alexandrino P, Branco M . (2010). A three-way contact zone between forms of Patella rustica (Mollusca: Patellidae) in the central Mediterranean Sea. Biol J Linn Soc 100: 154–169.
Sasa MM, Chippindale PT, Johnson NA . (1998). Patterns of postzygotic isolation in frogs. Evolution 52: 1811–1820.
Schilthuizen M, MCWG Giesbers, Beukeboom LW . (2011). Haldane’s rule in the 21st century. Heredity 107: 95–102.
Seehausen O, Takimoto G, Roy D, Jokela J . (2008). Speciation reversal and biodiversity dynamics with hybridization in changing environments. Mol Ecol 17: 30–44.
Sequeira F, Sodre D, Ferrand N, Bernardi JAR, Sampaio I, Schneider H et al. (2011). Hybridization and massive mtDNA unidirectional introgression between the closely related Neotropical toads Rhinella marina and R. schneideri inferred from mtDNA and nuclear markers. BMC Evol Biol 11: 264.
Simões PI, Lima AP, Farias IP . (2012). Restricted natural hybridization between two species of litter frogs on a threatened landscape in southwestern Brazilian Amazonia. Conserv Genet 13: 1145–1159.
Smadja CM, Butlin RK . (2011). A framework for comparing processes of speciation in the presence of gene flow. Mol Ecol 20: 5123–5140.
Sobel JM, Chen GF, Watt LR, Schemske DW . (2010). The biology of speciation. Evolution 64: 295–315.
Stöck M, Moritz C, Hickerson M, Frynta D, Dujsebayeva T, Eremchenko V et al. (2006). Evolution of mitochondrial relationships and biogeography of Palearctic green toads (Bufo viridis subgroup) with insights in their genomic plasticity. Mol Phylogenet Evol 41: 663–689.
Stöck M, Sicilia A, Belfiore N, Buckley D, Lo Brutto S, Lo Valvo M et al. (2008a). Post-Messinian evolutionary relationships across the Sicilian channel: Mitochondrial and nuclear markers link a new green toad from Sicily to African relatives. BMC Evol Biol 8: 56.
Stöck M, Roth P, Podloucky R, Grossenbacher K . (2008b). Wechselkröten–unter Berücksichtigung von Bufo viridis virdis Laurenti, 1768; Bufo variabilis (Pallas, 1769); Bufo boulengeri Lataste, 1879; Bufo balearicus Böttger, 1880 und Bufo siculus Stöck, Sicilia, Belfiore, Lo Brutto, Lo Valvo und Arculeo, 2008. In: Grossenbacher K, (ed) Handbuch der Amphibien und Reptilien Europas vol. 5 (Froschlurche II), AULA-Verlag: Wiesbaden. pp 413–498.
Stöck M, Savary R, Betto-Colliard C, Biollay S, Jourdan-Pineau H, Perrin N . (2013). Low rates of XY recombination, not turnovers, account for homomorphic sex chromosomes in several diploid species of Paleartic green toads (Bufo viridis subgroup). J Evol Biol 3: 674–682.
Sumida M, Kotaki M, Islam MM, Djong TH, Igawa T, Kondo Y et al. (2007). Evolutionary relationships and reproductive isolating mechanisms in the rice frog (Fejervarya limnocharis) species complex from Sri Lanka, Thailand, Taiwan and Japan, inferred from mtDNA gene sequences, allozymes, and crossing experiments. Zool Sci 24: 547–562.
Szymura JM . (1993). Analysis of hybrid zones with Bombina. In: Harrison R, (ed) Hybrid Zones and the Evolutionary Process. Oxford University Press: New York. pp 261–289.
Szymura J, Barton N . (1986). Genetic analysis of a hybrid zone between the fire-bellied toads, Bombina bombina and Bombina variegata, near Cracow in southern Poland. Evolution 40: 1141–1159.
Van Oosterhout C, Hutchinson WF, Derek P, Wills M, Shipley P . (2004). MicroChecker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Verardi A, Canestrelli D, Nascetti G . (2009). Nuclear and mitochondrial patterns of introgression between the parapatric European treefrogs Hyla arborea and H. intermedia. Annls Zool Fennici 46: 247–258.
Vines TH, Köhler SC, Thiel M, Ghira I, Sands TR, MacCullum CJ et al. (2003). The maintenance of reproductive isolation in a mosaic hybrid zone between the fire-bellied toads Bombina bombina and B. variegata. Evolution 57: 1876–1888.
Wilson AC, Maxson LR, Starich VM . (1974). Two types of molecular evolution. Evidence from studies of interspecific hybridization. Proc Natl Acad Sci USA 71: 2843–2847.
Woolfit M, Bromham L . (2005). Population size and molecular evolution on islands. Proc R Soc B 272: 2277–2282.
Acknowledgements
This work was partly supported by a Heisenberg-Fellowship (Sto 493/2-1), and grant Sto 493/3-1 of the German Science Foundation (DFG), as well as in part by the Fondation Agassiz of the University of Lausanne (1 July 2010)—all to MS; by funds to NP (Swiss National Science Foundation, grant 31003A-129894), CD (PhD fellowship from the Faculty of Biology and Medicine of the University of Lausanne), and the Italian Ministry of the University (PRIN 20085YJMTC) to LB. We thank M Benà, L Bedin, S Bertollo, E Boschetti, E Cirani, F di Montereale, M Dovigo, B Favaretto, GF Ficetola, B Golfieri, S Mazzotti, D Miserocchi, MG Mitri, L Ometto, R Pollo, P Reggiani, J Richard, E Romanazzi, M Semenzato, M Simonazzi, GF Turrisi, M Uliana, for contributing samples and/or helping with fieldwork, L Ometto and A Sicilia for initiating some of the contacts leading to our collaboration, and AS for help in the lab; D Pio, O Broennimann and S Antoniazza for discussions and three anonymous reviewers for helpful comments on previous versions of this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article
Cite this article
Dufresnes, C., Bonato, L., Novarini, N. et al. Inferring the degree of incipient speciation in secondary contact zones of closely related lineages of Palearctic green toads (Bufo viridis subgroup). Heredity 113, 9–20 (2014). https://doi.org/10.1038/hdy.2014.26
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.26
This article is cited by
-
Easternmost distribution of Bufo bufo (Linnaeus, 1758) in Türkiye: implications for the putative contact zone between B. bufo and B. verrucosissimus
Genetica (2023)
-
Genetic Divergence Across Glacial Refugia Despite Interglacial Gene Flow in a Crested Newt
Evolutionary Biology (2021)
-
The effect of phylogeographic history on species boundaries: a comparative framework in Hyla tree frogs
Scientific Reports (2020)
-
Phylogeography of Aegean green toads (Bufo viridis subgroup): continental hybrid swarm vs. insular diversification with discovery of a new island endemic
BMC Evolutionary Biology (2018)
-
The absence of reproductive isolation between non-sister and deeply diverged mitochondrial lineages of the black-throated tit (Aegithalos concinnus) revealed by a multilocus genetic analysis in a contact zone
BMC Evolutionary Biology (2017)
