
Abstract
Sex determination mechanisms (SDMs) show striking diversity and appear to evolve rapidly. Although interspecific comparisons and studies of ongoing major transitions in sex determination (such as the establishment of new sex chromosomes) have shed light on how SDMs evolve, comparatively little attention has been paid to intraspecific variation with less drastic effects. In this study, I used mutant strains carrying a temperature-sensitive sex determination mutation, along with a second null mutation, in different wild genetic backgrounds to uncover hidden variation in the SDM of the model nematode Caenorhabditis elegans. I then used quantitative trait locus (QTL) mapping to begin to investigate its genetic basis. I identified several QTLs, and although this variation apparently involved genotype-by-temperature interactions, QTL effects were generally consistent across temperatures. These QTLs collectively and individually explained a relatively large fraction of the variance in tail morphology (a sexually dimorphic trait), and two QTLs contained no genes known to be involved in somatic sex determination. These results show the existence of within-species variation in sex determination in this species, and underscore the potential for microevolutionary change in this important developmental pathway.
Similar content being viewed by others

Introduction
Although reproduction with two distinct sexes is taxonomically widespread, the mechanisms that determine an individual's sex are remarkably diverse across species. At the highest level, sex determination mechanisms (SDMs) can be divided into two major categories: those in which the organism's sex is determined by its genotype (genotypic sex determination), and those in which sex is determined by some environmental input during development (environmental sex determination). There is great variation within these categories in the most upstream determiner of sex. There are genotypic sex determination systems, for instance, relying on different types of sex chromosomes as in mammals and birds (Haag and Doty, 2005), systems in which sex determination seems to be influenced by many genes (Vandeputte et al., 2007), and systems in which one sex is haploid and the other diploid (Cook and Crozier, 1995). Similarly, environmental sex determination systems use an array of different environmental variables, including population density and host crowding (Blackmore and Charnov, 1989), light availability (Korpelainen, 1998), and perhaps most commonly, temperature (temperature-dependent sex determination; Janzen and Phillips, 2006).
Within these types of SDMs lies even more diversity, in the genetic pathways that translate the upstream sex-determining factor into a signal triggering male or female development. Many dipterans, for example, possess an XX/XY sex chromosome system (Sanchez, 2008), but although the most downstream gene in this pathway, doublesex, is conserved, there are important differences among species elsewhere in the pathway. For example, sex-specific splicing of tra is regulated by Sxl in Drosophila (Cline and Meyer, 1996) but is directed by tra itself in an auto-regulatory feedback loop in Ceratitis (Pane et al., 2002) and Musca (Hediger et al., 2010), with Sxl lacking a sex-specific function in both of these species (Meise et al., 1998; Saccone et al., 1998).
From these types of interspecific pathway comparisons as well as phylogenetic and theoretical studies, we gained some key insights into the evolution of SDMs. First, changes in SDMs may occur commonly and easily, at least in some clades. For example, different genotypic sex determination and temperature-dependent sex determination mechanisms are phylogenetically scattered in reptiles (Janzen and Phillips, 2006) and teleost fishes (Mank et al., 2006), indicating frequent and independent transitions. Second, genes involved in sex determination evolve rapidly and often coevolve with one another. This conclusion is supported by huge sequence divergence among related species in these genes (Whitfield et al., 1993; de Bono and Hodgkin, 1996) and functional analyses (Haag et al., 2002). Finally, changes to SDMs typically occur in the upstream portions of the pathway (Wilkins, 1995; Pomiankowski et al., 2004), as these show more divergence among species, whereas downstream components are more conserved (Raymond et al., 1998).
However, to appreciate more fully how SDMs change over time, an understanding of both between-species divergence and within-species variation is necessary. Although earlier work on intraspecific variation and the microevolution of SDMs (Bull and Charnov, 1977) was largely theoretical and abstract because of a lack of molecular understanding of most SDMs (Pomiankowski et al., 2004), great strides are rapidly being made in this area. For example, Hediger et al. (2010) recently showed that gain-of-function alleles of the Musca domestica tra ortholog correspond to the dominant feminizing factors segregating in some populations. Similarly, various XX/XY and ZZ/ZW SDMs involving LG (linkage group)5 and LG7 occur in different species of Lake Malawi cichlids, which have diversified only in the last million years; in some cases, sex is even determined by an epistatic interaction between LG5 and LG7 (Ser et al., 2010). The maintenance or fixation of these types of sex-determining variants is typically attributed to various types of selection (Kozielska et al., 2009; Roberts et al., 2009).
One commonality in these and other cases of intraspecific variation in SDMs (Kallman, 1968; Ogata et al., 2008) is that they involve variants with major effects, such as novel sex-determining genes or chromosomes, or taxa in which related species show very different SDMs (for example, medakas; Takehana et al., 2008). Thus, there is evidence that SDM evolution is often characterized by major and rapid changes. However, what about species not currently experiencing such extreme changes; are they in complete stasis or do SDMs change gradually as well? Intraspecific SDM variation of smaller effect has received comparatively little attention, perhaps because of an ascertainment bias toward variants with drastic effects. However, gradual or cumulative change should not be dismissed completely just yet. For example, heritable quantitative variation has been shown to occur in sex ratio reaction norms in several species with environmental sex determination (Blackmore and Charnov, 1989; Rhen and Lang, 1998), although its genetic basis is unknown and effective heritability in the wild may be low. In addition, minor-effect alleles may be masked in genotypic sex determination species if SDMs show some degree of genetic robustness, allowing the underlying pathway to evolve (True and Haag, 2001). In fact, other developmental signaling pathways have been shown to evolve even without any change in the final phenotypic output, such as the signaling network underlying vulval development in nematodes (Félix, 2007; Zauner and Sommer, 2007; Milloz et al., 2008). Moreover, strong alleles may not necessarily be the result of a single mutation, but rather assembled cumulatively by standing variation and successive minor-effect mutations (Rebeiz et al., 2009). Finally, certain patterns in SDM evolution may not be fully explained by this model of swift and significant changes in master sex determiners. For example, coevolution among interacting sex-determining genes, even intermediate and downstream genes, occurs without changes in the roles of the genes, their interaction or in the master sex-determining signal itself (Haag et al., 2002).
In this study, I investigated variation in sex determination in Caenorhabditis elegans, a model nematode ‘worm’. This is an ideal study species because its sex determination cascade has been well characterized by years of study (reviewed in Wolff and Zarkower, 2008). In wild-type worms, sex is determined by sex chromosomes; males are XO, whereas hermaphrodites, which are essentially females that produce a limited supply of sperm early in development, are XX. By examining the joint quantitative effects of specific mutations in two key sex determination genes (Chandler et al., 2009) in different genetic backgrounds, I uncovered a cryptic intraspecific variation in the sex determination pathway. I then used quantitative trait locus (QTL) mapping to begin to investigate its underlying genetic basis and microevolutionary potential, with the goal of addressing the following questions: (1) How many loci are involved, and what are the magnitudes of their effects? (2) Do any QTLs lack candidate genes with known roles in sex determination? and (3) Do QTLs influence overall pathway activity, with effects consistent across environments (in this case, temperatures), or are they more complex, with genotype-by-environment interactions? I conclude by discussing the implications of these findings for our understanding of how SDMs evolve.
Materials and methods
Strains and strain construction
Strain CB5362 tra-2(ar221ts)II; xol-1(y9)X (Hodgkin, 2002; Chandler et al., 2009) consists of XX worms, which are primarily hermaphrodites at permissive temperatures (<∼16 °C) and males at warmer temperatures (>∼20 °C). At intermediate temperatures, tail phenotypes are variably intersexed (Figure 1). This strain was originally constructed with the N2 genetic background (Brenner, 1974). I introgressed these two mutations into the genetic backgrounds of four additional wild isolates, chosen for their molecular, phenotypic and geographic diversity (Milloz et al., 2008): CB4856 from Hawaii, MY2 from Germany, AB1 from Australia and JU258 from Madeira (Hodgkin and Doniach, 1997; Barriere and Félix, 2005; Haber et al., 2005).

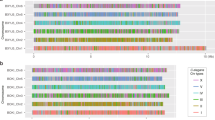
Scoring system for tail morphology. 1=wild-type hermaphrodite; 2=tail whip is present but shortened or masculinized; 3=tail whip is absent but tail retains overall hermaphrodite shape ; 4=tail has blunted, male-like shape but lacks fan and rays; 5=fan or rays present but reduced in size or number; 6=wild-type male. A full color version of this figure is available at the Heredity Journal online.
I backcrossed the tra-2(ar221) and xol-1(y9) alleles into each genetic background separately, selecting for each mutation with a combination of molecular and phenotypic markers, for 10 generations with CB4856, and for 4 generations with MY2, AB1 and JU258. After backcrossing, I crossed the tra-2 and xol-1 introgression lines, selfed the offspring and selected double mutants with each new genetic background.
I performed QTL mapping using the temperature-sensitive lines with N2 and the CB4856 genetic backgrounds, as they display many single-nucleotide polymorphisms and are commonly used for mapping (Gutteling et al., 2007; Seidel et al., 2008). To construct recombinant inbred lines (RILs), which are available from the author upon request, for QTL mapping, I first performed reciprocal crosses between these strains. I picked F1 offspring at 13 °C, allowed them to self-fertilize and oviposit and used a PCR marker flanking an indel polymorphism on chromosome III to verify heterozygosity of cross-progeny. I picked F2 offspring to individual plates, subsequently transferring 1–4 offspring to new plates for a total of 10 generations. At the F10 generation, RILs were grown to large population sizes to obtain working cultures and frozen stocks.
Phenotypic assays
I scored each temperature-sensitive strain as well as wild-type male and hermaphrodite N2 worms as controls, for both gonadal sex ratio and tail phenotype at four rearing temperatures (24, 18, 16 and 13 °C), using standard bleaching protocols to obtain age-synchronized populations (with approximately 150–200 worms each) for assays. At adulthood, I mounted worms onto glass slides (typically containing 20–50 worms per slide) with 25 mM NaN3 for Nomarski microscopy, using standard methods (Shaham, 2006). I photographed all worms using a Zeiss Axioplan II (Zeiss, Thornwood, NY, USA) with differential interference contrast optics within a few hours of mounting.
I cropped individual worms from digital images and scored them for gonadal sex and tail phenotypes blindly with regard to strain and rearing temperature. Gonadal sex was scored as a binary trait (0=hermaphrodite, 1=male). Hermaphrodites were identified by their distinctive two-armed gonad morphology and/or the presence of oocytes within the adult worm, whereas males were recognized by their one-armed gonads and lack of oocytes. Worms with gonads that were abnormal, unclear or ambiguous (∼5% of the total) were excluded from sex ratio analyses. Tail morphology was scored on a semi-quantitative scale from 1 to 6 (Figure 1). A score of 1 was given to worms indistinguishable from wild-type hermaphrodites, with a fully developed tail spike. A score of 2 was assigned to worms with an abnormal but still hermaphrodite-like tail, indicated by a truncated (but still present) tail spike and/or slight swelling at the base of the tail. Worms with more intersexed tail morphology, lacking a tail spike altogether but also lacking a flattened male shape, were given a score of 3. A score of 4 was assigned to individuals with a more male-like tail shape, but without the fan and rays characteristic of normal male tails. A score of 5 was given to worms having a male-shaped tail with at least some of the male characteristics such as a fan or rays, but in which these features were reduced in number or size. A score of 6 was reserved for worms with tails indistinguishable from those of wild-type males, complete with full fan, rays and spicules. To confirm the repeatability of the rating scale, I blindly rescored a subset of worms and calculated Cohen's κ with quadratic weightings, a statistic commonly used to measure repeatability in ordinal ratings (Banerjee et al., 1999).
Genotyping
I washed worms off stock cultures in deionized water into 1.5 ml tubes, centrifuged them, removed the supernatant and froze the pelleted samples at −80 °C. I extracted DNA from thawed tissue samples using a Qiagen DNeasy Blood and Tissue kit according to the manufacturer's directions (Qiagen, Valencia, CA, USA). DNA concentrations were quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA), and samples were diluted to 10 ng μl−1. RIL genotyping at 90 single-nucleotide polymorphisms (Supplementary Table S1) was performed using the Sequenom MassARRAY system (Sequenom, San Diego, CA, USA) at the Iowa State University Genomic Technologies Facility.
Analyses
To test the effects of rearing temperature and genetic background on gonadal sex ratio in CB5362 and the introgression lines, I compared nested models using hierarchical likelihood ratio tests. For sex ratio, I evaluated a series of logistic models to test the effects of temperature, strain and strain-by-temperature interaction. For tail morphology, I used ordered logistic models, an extension of logistic models allowing for more than two categories in the response variable, to test the effects of temperature, strain and two different strain-by-temperature interactions. Each ordered logistic model was composed of a series of five parallel logistic models (because there are six possible tail scores), with each logistic model j expressing the probability that an individual's tail score is greater than j. Maximum likelihood values and parameter estimates for each model were obtained using the ‘optim’ function in R v2.10.0 (R Development Core Team) using the BFGS (Broyden–Fletcher–Goldfarb–Shanno) search algorithm.
I performed QTL mapping using the software package R/qtl v1.14-2 in R (Broman et al., 2003), using gonadal sex ratio and tail scores each at four rearing temperatures. To look for QTLs with effects on reaction norm shape, I also performed QTL analyses on tail reaction norm slopes from 13 to 16 °C, and from 16 to 18 °C. Strain means were used as the phenotypic values. I first checked the genotype data for evidence of segregation distortion and disagreement with the publicly available genetic map (WormBase web site, http://www.wormbase.org, release WS208). I performed an initial exploratory QTL analysis by interval mapping. Subsequently, I used the ‘stepwiseqtl’ function to search for additive QTLs and compare multiple-QTL models. The latter approach compares models using logarithm of odds (LOD) scores, imposing a penalty for the inclusion of additional QTLs, and is advantageous because it allows one to test the effects of multiple QTLs simultaneously, offering increased detection power (Broman and Sen, 2009). Penalties were computed from 1000 permutations of the ‘scantwo’ function. For each QTL, 1.5-LOD confidence intervals were estimated using the ‘lodint’ function, and effect estimates and the proportion of variance explained were both estimated using ‘fitqtl’. All analyses were conducted using multiple imputation with 64 draws.
Results
Variation among genetic backgrounds
A total of 711 worms from CB5362 and the 4 introgression lines, and 9336 worms from the RIL population were examined (average n=∼35 for each temperature by strain combination). All control hermaphrodite N2 worms were assigned tail scores of 1, and all male N2 worms were given tail scores of 6. Repeatability of tail scores was very high (weighted κ=0.97, n=368).
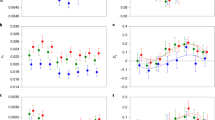
The ‘Temp+Strain’ model best explained variation in the gonadal sex ratio (Table 1; Figure 2), indicating that sex ratio reaction norms varied among strains in their horizontal positions, but not shapes. The ‘Temp+Strain+I1+I2’ model was selected as the best to explain variation in tail scores (Table 1; Figure 2), suggesting that tail morphology reaction norms in different strains were not simply horizontally displaced as a whole, but rather that reaction norm shape and the distribution of tail scores differed among strains as well, indicating a temperature-by-strain interaction.

Plots of (a) gonadal sex ratio and (b) mean tail morphology score as a function of rearing temperature for tra-2(ar221); xol-1(y9) strains with different genetic backgrounds.
QTL analyses
In total, 157 RILs were generated; a subset of 66 was both phenotyped (Figure 3) and genotyped (Supplementary Table S2); of the 90 original markers, 66 were informative. The range of phenotypes in the RILs exceeded that of the parental lines, except at 24 °C (Figure 3). A predominance of RILs with more feminized phenotypes, particularly at 18 °C, suggests there may have been unintentional selection during RIL construction. Although there were no apparent problems in the genetic map estimated from the genotype data, there were a few regions showing some segregation distortion (Table 2). One region of very strong segregation distortion on chromosome I probably resulted from incompatibility between the N2 and CB4856 alleles at the zeel-1 and peel-1 loci (Seidel et al., 2008). Regions of modest segregation distortion on chromosomes III, IV and V may have been the result of inadvertent selection for hermaphrodites during RIL construction, as these regions all contained QTLs, and the overrepresented allele in each case was associated with more feminized phenotypes. Two additional markers on chromosome III and one on chromosome II showing distortion were not closely linked to any QTL but may have been the result of genotyping errors, indicated by the relatively large number of individuals missing genotypes at these markers.

Plots of (a) gonadal sex ratio and (b) mean tail morphology score as a function of rearing temperature for recombinant inbred lines. The parental lines are shown in bold.
A total of six distinct main QTLs was identified by model selection (Table 3). The only trait for which all of these QTLs were evident simultaneously was the tail score at 16 °C. However, the QTL on chromosome IV was also evident for the tail phenotype at 13 and 18 °C and for sex ratio at 16 °C, and the QTL on chromosome I and one of the QTLs on chromosome V were also detected for the tail phenotype at 13 °C. No QTLs were detected for the tail score at 24 °C, sex ratio at 13 °C or 24 °C or for either measure of tail reaction norm shape.
The QTLs identified for sex ratio at 18 °C (Table 3) are probably statistical artifacts, for several reasons. The confidence interval for the QTL on chromosome I for this trait is very narrow and is entirely within the region of strong segregation distortion corresponding to the zeel-1/peel-1 incompatibility (Table 2). Second, the two QTLs on chromosome IV are closely linked, in fact with overlapping confidence intervals, and are in repulsion; such a pattern can often provide a strong but spurious model fit, and therefore it is best not to consider models with more than one QTL between any two adjacent markers, as in this case (Broman and Sen, 2009). Finally, no models considering these three QTLs individually or in pairwise combinations performed better than the null model (all penalized LOD scores <0).
Although not all these QTLs are obvious in LOD profiles from standard interval mapping (Figure 4), this is expected because of the increased power of multiple-QTL models, as they test the effects of each putative QTL while accounting for variation due to other QTLs (similar to composite interval mapping). This scenario is particularly likely for my data, because one QTL (on chromosome IV) had very large effects that would obscure the signal for the QTLs of modest effect during interval mapping. Importantly, composite interval mapping (Wang et al., 2007) produced similar results and did not affect overall conclusions, suggesting that my findings are robust to the particular choice of statistical models for QTL mapping (for a discussion of the differences between these two methods, see Broman and Sen, 2009).

LOD plots from exploratory interval mapping (IM) analysis of tail morphology and sex ratio at four rearing temperatures and of tail reaction norm slope in two temperature intervals, showing overall qualitative similarity for sex ratio and tail phenotypes across temperatures. Horizontal dashed lines indicate significance thresholds for IM computed by permutation. Sex ratio at 13 °C is omitted because there was no variation in sex ratio among RILs at that temperature. QTL significance (Table 3) was then assessed using a more powerful multiple-QTL model selection approach.
Discussion
To understand how SDMs evolve, we need to investigate variation in these pathways within species, in addition to divergence among species. Most previous studies on SDM microevolution have focused on individual or closely related groups of species segregating alleles with extremely large effects, such as loss-of-function alleles or dominant male- or female-determining factors, and which may be undergoing major transitions to new SDMs (Hediger et al., 2010; Ser et al., 2010). In contrast, in this study, I characterized variation in the SDM of C. elegans, an important model organism which is unlikely to be currently experiencing such a radical transition.
My results show that the joint effects of the tra-2(ar221) and xol-1(y9) alleles on gonadal sex ratio and sexually dimorphic tail phenotypes vary subtly but significantly with the wild genetic background in which they occur. Moreover, the discovery of at least one QTL influencing both gonadal sex and tail morphology (Table 3; Figure 4) supports the idea that at least some of this variation is related to sex determination itself and not, say, genes influencing the differentiation of sex-specific tail phenotypes after sex has been determined. Interestingly, many RILs showed more extreme phenotypes than either parental line, similar to the results of Zauner and Sommer (2007). This pattern of transgressive segregation is not uncommon (Rieseberg et al., 1999, 2003) and can be explained by, among other mechanisms, the co-occurrence of antagonistic QTL alleles within each parental line, which then recombine to produce more extreme phenotypes in the F2 and later generations. In this case, the transgressive phenotypes were biased in one direction (Figure 3), specifically toward hermaphrodite-skewed sex ratios and feminized tail morphology. This bias may reflect an unintentional selection for less masculinized hermaphrodites during RIL construction, consistent with patterns of segregation distortion at markers linked to QTL (Tables 2 and 3).
The presence of a genotype-by-environment (G × E; in this case, the environmental variable is temperature) interaction for tail phenotype (Table 1; Figure 2) suggests that this variation is not merely due to differences in overall pathway activity, but rather due to variation in how sex-determining genes are regulated. In addition, although variation in transcription levels of sex-determining genes has been documented in Drosophila melanogaster (Tarone et al., 2005), translational and posttranslational regulation both have crucial roles in sex determination in C. elegans (Clifford et al., 2000; Sokol and Kuwabara, 2000) and therefore may be important components of this variation. QTL mapping showed a small handful of loci underlying this variation, individually explaining moderate to large proportions (up to ∼50%) of the among-strain variance in tail morphology and sex ratio at any particular temperature (Table 3). Thus, although this variation in C. elegans is less obvious compared with other species with known SDM polymorphisms, as it is only revealed in the context of a mutant genotype, its genetic basis is probably not too complex to dissect further with high-resolution mapping and candidate gene studies.
Some QTLs had apparent effects at multiple temperatures. Although finer mapping is necessary to completely exclude the possibility that the seemingly common QTLs at different temperatures are distinct but closely linked, this observation suggests that these QTLs have effects that are generally consistent across environments, particularly the QTL with the largest effects, and therefore may reflect differences in overall pathway activity. Other QTLs had effects that were temperature-specific, suggesting possible regulatory differences among these genetic backgrounds. However, qualitative similarity of LOD profiles across temperatures for both tail morphology and sex ratio (Figure 4) is also suggestive of common effects that were simply too weak to be detected in some cases. Moreover, despite the presence of G × E interactions among the five genetic backgrounds (Table 1) and apparently substantial variation for reaction norm slope between 16 and 18 °C (Figure 3) among the RILs, no QTLs influencing the reaction norm slope were detected. This finding implies that the genetic architecture of the reaction norm shape may differ from that of the reaction norm intercept (and hence overall pathway activity or expression), perhaps distributed across more loci with effects too small to be identified in this study. Whether this conclusion applies to other reaction norms (such as thermal sex ratio reaction norms in reptiles with temperature-dependent sex determination) remains to be tested. In contrast, however, several QTL mapping studies of phenotypic plasticity and G × E interactions in both morphological and life history traits have detected QTLs influencing reaction norm slopes and with environment-specific effects (Gurganus et al., 1998; Vieira et al., 2000; Gutteling et al. 2007).
What conclusions can we draw from these findings? First, even if a species is not currently experiencing a radical transition between SDMs, such as the fixation of a novel male-determining factor or the conversion of an autosome to a sex chromosome, it may still harbor functional variation in its SDM. This observation suggests that in addition to major transitions in upstream sex-determining genes, SDMs may also be subjected to more gradual change in other parts of the pathway, for example, in the relative importance of redundant gene interactions. In C. elegans, for instance, tra-2 regulates tra-1 by two distinct manners: directly and through the fem genes; both are required for hermaphrodite spermatogenesis (Lum et al., 2000). In Caenorhabditis briggsae, although, which evolved self-fertility independently, indirect regulation through fem intermediates is dispensable for spermatogenesis in hermaphrodites (Hill et al., 2006). Moreover, both fem-3 and fem-2 are epistatic to tra-1 in the C. elegans germ line, but only fem-3 is in C. briggsae (Hill and Haag, 2009). Thus, these species have taken different paths to the same trait. Such observations have led to the proposal that the differential loss and strengthening of ancestral redundant gene interactions in separate lineages (driven by either drift or, probably in the case of this example, selection) caused some of the interspecific differences in SDMs that we see today (Ellis, 2006). Sex determination in C. elegans must involve such previously unknown, and probably minor or redundant, factors, and at least some of these are variable within this species. For example, two of the QTLs (on chromosomes I and V) influencing somatic sexual phenotypes contained no candidate genes with a known role in somatic sex determination (Table 3). It will be interesting to see whether these minor components are shared by other Caenorhabditis species; it is worth noting that qualitative similarities have been discovered between intraspecific and interspecific variation in the signaling pathways underlying Caenorhabditis vulva patterning (Félix, 2007; Milloz et al., 2008) and in the basis of sexually dimorphic abdominal coloration in Drosophila (Kopp et al., 2003).
In addition, the presence of this variation may help explain the coevolutionary change seen in interacting sex-determining genes. Coevolution is expected if amino-acid substitutions altering the function of one gene help promote the fixation of compensatory mutations in its interaction partners (Haag et al., 2002). Although polymorphism in the sex-determining genes that have been examined in C. elegans is low (Graustein et al., 2002; Haag and Ackerman, 2005), to illustrate this possibility, there is one nonsynonymous single-nucleotide polymorphism in tra-3, the function of which in sex determination is to activate TRA-2A protein by cleavage (Sokol and Kuwabara, 2000), and which is an excellent candidate for the strongest QTL identified in this study (Table 3). If this polymorphism affects the affinity of TRA-3 for TRA-2A and is responsible for selectable phenotypic variation, it could favor compensatory changes in TRA-2A structure. Interestingly, this single-nucleotide polymorphism mediates the influence of temperature on body size in C. elegans (Kammenga et al., 2007), suggesting that the initial changes to sex-determining genes might have pleiotropic effects that could promote their fixation.
Along those lines, the selective forces influencing the maintenance or fixation of these variants in SDMs are an important consideration. Previous theoretical and empirical work has suggested that sex ratio selection (Bulmer and Bull, 1982; Conover and Van Voorhees, 1990; Wilkins, 1995), sexual selection and sexual conflict (Pomiankowski et al., 2004; van Doorn and Kirkpatrick, 2007), as well as parent–offspring conflict (Werren et al., 2002) can drive the evolution of SDMs. This and other recent work examining the ‘cryptic’ evolution of developmental signaling pathways may offer additional hypotheses. Milloz et al. (2008) hypothesized that cryptic variation among strains of C. elegans in cell signaling related to vulva patterning (1) could be neutral, (2) might not be cryptic at all, but phenotypically exposed to selection in different environments or (3) may be a correlated response to selection on other traits affected by the same signaling pathways. Zauner and Sommer (2007) proposed that variation in cell–cell interactions involved in vulval patterning within and among Pristionchus species might be due to selection for robustness to developmental noise in different environments, similar to the second hypothesis mentioned above. These possibilities likely apply to this SDM variation as well. For example, many ‘core’ sex-determining genes have secondary roles in dosage compensation, meiosis, etc. (for example, gld-1; Francis et al., 1995; Clifford et al., 2000). Moreover, this type of cryptic variation may have evolutionary consequences, for example, on evolvability (de Visser et al., 2003), and SDM and sex chromosome evolution in particular may contribute to post-zygotic reproductive isolation (Wade et al., 1997; Baird, 2002).
This study illustrates how laboratory-generated mutations can be exploited to expose hidden variation in, and thus explore the microevolution of, sex determination, even in species that appear to lack such variation at first glance. I have shown that even taxa not currently undergoing major transitions in SDMs may harbor variation in sex determination, and that such hidden variation is thought to lead eventually to pathway divergence. Further investigation of the QTL identified in this study, such as fine mapping to pinpoint the causative genes and more detailed cellular analyses of phenotypes, should provide a better view of their molecular bases. Examining how these variants interact with one another, and whether they have any phenotypic effects in nonmutant genotypes, will also illuminate what selective forces, if any, influence their evolution.
References
Baird SE (2002). Haldane's rule by sexual transformation in Caenorhabditis. Genetics 161: 1349–1353.
Banerjee M, Capozzoli M, McSweeney L, Sinha D (1999). Beyond Kappa: a review of interrater agreement measures. Can J Stat 27: 3–23.
Barriere A, Félix MA (2005). High local genetic diversity and low outcrossing rate in Caenorhabditis elegans natural populations. Curr Biol 15: 1176–1184.
Blackmore MS, Charnov EL (1989). Adaptive variation in environmental sex determination in a nematode. Am Nat 134: 817–823.
Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77: 71–94.
Broman KW, Sen S (2009). A Guide to QTL Mapping with R/qtl. Springer: New York, NY.
Broman KW, Wu H, Sen S, Churchill GA (2003). R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890.
Bull JJ, Charnov EL (1977). Changes in heterogametic mechanism of sex determination. Heredity 39: 1–14.
Bulmer MG, Bull JJ (1982). Models of polygenic sex determination and sex-ratio control. Evolution 36: 13–26.
Chandler CH, Phillips PC, Janzen FJ (2009). The evolution of sex-determining mechanisms: lessons from temperature-sensitive mutations in sex determination genes in Caenorhabditis elegans. J Evol Biol 22: 192–200.
Clifford R, Lee MH, Nayak S, Ohmachi M, Giorgini F, Schedl T (2000). FOG-2, a novel F-box containing protein, associates with the GLD-1 RNA binding protein and directs male sex determination in the C. elegans hermaphrodite germline. Development 127: 5265–5276.
Cline TW, Meyer BJ (1996). Vive la différence: males vs females in flies vs worms. Annu Rev Genet 30: 637–702.
Conover DO, Van Voorhees DA (1990). Evolution of a balanced sex ratio by frequency-dependent selection in a fish. Science 250: 1556–1558.
Cook JM, Crozier RH (1995). Sex determination and population biology in the Hymenoptera. Trends Ecol Evol 10: 281–286.
de Bono M, Hodgkin J (1996). Evolution of sex determination in Caenorhabditis: unusually high divergence of tra-1 and its functional consequences. Genetics 144: 587–595.
de Visser JAGM, Hermisson J, Wagner GP, Meyers LA, Bagheri HC, Blanchard JL et al. (2003). Perspective: evolution and detection of genetic robustness. Evolution 57: 1959–1972.
Ellis RE (2006). Enigma variations: control of sexual fate in nematode germ cells. Genome Biol 7: 227.
Félix MA (2007). Cryptic quantitative evolution of the vulva intercellular signaling network in Caenorhabditis. Curr Biol 17: 103–114.
Francis R, Maine E, Schedl T (1995). Analysis of the multiple roles of gld-1 in germline development: interactions with the sex determination cascade and the glp-1 signaling pathway. Genetics 139: 607–630.
Graustein A, Gaspar JM, Walters JR, Palopoli MF (2002). Levels of DNA polymorphism vary with mating system in the nematode genus Caenorhabditis. Genetics 161: 99–107.
Gurganus MC, Fry JD, Nuzhdin SV, Pasyukova EG, Lyman RF, Mackay TFC (1998). Genotype-environment interaction at quantitative trait loci affecting sensory bristle number in Drosophila melanogaster. Genetics 149: 1883–1898.
Gutteling EW, Riksen JAG, Bakker J, Kammenga JE (2007). Mapping phenotypic plasticity and genotype-environment interactions affecting life-history traits in Caenorhabditis elegans. Heredity 98: 28–37.
Haag ES, Ackerman AD (2005). Intraspecific variation in fem-3 and tra-2, two rapidly coevolving nematode sex-determining genes. Gene 349: 35–42.
Haag ES, Doty AV (2005). Sex determination across evolution: connecting the dots. PLoS Biol 3: 21–24.
Haag ES, Wang SP, Kimble J (2002). Rapid coevolution of the nematode sex-determining genes fem-3 and tra-2. Curr Biol 12: 2035–2041.
Haber M, Schungel M, Putz A, Muller S, Hasert B, Schulenburg H (2005). Evolutionary history of Caenorhabditis elegans inferred from microsatellites: evidence for spatial and temporal genetic differentiation and the occurrence of outbreeding. Mol Biol Evol 22: 160–173.
Hediger M, Henggeler C, Meier N, Perez R, Saccone G, Bopp D (2010). Molecular characterization of the key switch F provides a basis for understanding the rapid divergence of the sex-determining pathway in the housefly. Genetics 184: 155–170.
Hill RC, de Carvalho CE, Salogiannis J, Schlager B, Pilgrim D, Haag ES (2006). Genetic flexibility in the convergent evolution of hermaphroditism in Caenorhabditis nematodes. Dev Cell 10: 531–538.
Hill RC, Haag ES (2009). A sensitized genetic background reveals evolution near the terminus of the Caenorhabditis germline sex determination pathway. Evol Dev 11: 333–342.
Hodgkin J (2002). Exploring the envelope: systematic alteration in the sex-determination system of the nematode Caeraorhabditis elegans. Genetics 162: 767–780.
Hodgkin J, Doniach T (1997). Natural variation and copulatory plug formation in Caenorhabditis elegans. Genetics 146: 149–164.
Janzen FJ, Phillips PC (2006). Exploring the evolution of environmental sex determination, especially in reptiles. J Evol Biol 19: 1775–1784.
Kallman KD (1968). Evidence for the existence of transformer genes for sex in the teleost Xiphophorus maculatus. Genetics 60: 811–828.
Kammenga JE, Doroszuk A, Riksen JAG, Hazendonk E, Spiridon L, Petrescu AJ et al. (2007). A Caenorhabditis elegans wild type defies the temperature-size rule owing to a single nucleotide polymorphism in tra-3. PLoS Genet 3: 358–366.
Kopp A, Graze RM, Xu SZ, Carroll SB, Nuzhdin SV (2003). Quantitative trait loci responsible for variation in sexually dimorphic traits in Drosophila melanogaster. Genetics 163: 771–787.
Korpelainen H (1998). Labile sex expression in plants. Biol Rev 73: 157–180.
Kozielska M, Weissing FJ, Beukeboom LW, Pen I (2009). Segregation distortion and the evolution of sex-determining mechanisms. Heredity 104: 100–112.
Lum DH, Kuwabara PE, Zarkower D, Spence AM (2000). Direct protein-protein interaction between the intracellular domain of TRA-2 and the transcription factor TRA-1A modulates feminizing activity in C. elegans. Genes Dev 14: 3153–3165.
Mank JE, Promislow DEL, Avise JC (2006). Evolution of alternative sex-determining mechanisms in teleost fishes. Biol J Linnean Soc 87: 83–93.
Meise M, Hilfiker-Kleiner D, Dubendorfer A, Brunner C, Nothiger R, Bopp D (1998). Sex-lethal, the master sex-determining gene in Drosophila, is not sex-specifically regulated in Musca domestica. Development 125: 1487–1494.
Milloz J, Duveau F, Nuez I, Félix M-A (2008). Intraspecific evolution of the intercellular signaling network underlying a robust developmental system. Genes Dev 22: 3064–3075.
Ogata M, Hasegawa Y, Ohtani H, Mineyama M, Miura I (2008). The ZZ/ZW sex-determining mechanism originated twice and independently during evolution of the frog, Rana rugosa. Heredity 100: 92–99.
Pane A, Salvemini M, Bovi PD, Polito C, Saccone G (2002). The transformer gene in Ceratitis capitata provides a genetic basis for selecting and remembering the sexual fate. Development 129: 3715–3725.
Pomiankowski A, Nothiger R, Wilkins A (2004). The evolution of the Drosophila sex-determination pathway. Genetics 166: 1761–1773.
Raymond CS, Shamu CE, Shen MM, Seifert KJ, Hirsch B, Hodgkin J et al. (1998). Evidence for evolutionary conservation of sex-determining genes. Nature 391: 691–695.
Rebeiz M, Pool JE, Kassner VA, Aquadro CF, Carroll SB (2009). Stepwise modification of a modular enhancer underlies adaptation in a Drosophila population. Science 326: 1663–1667.
Rhen T, Lang JW (1998). Among-family variation for environmental sex determination in reptiles. Evolution 52: 1514–1520.
Rieseberg LH, Archer MA, Wayne RK (1999). Transgressive segregation, adaptation and speciation. Heredity 83: 363–372.
Rieseberg LH, Widmer A, Arntz AM, Burke JM (2003). The genetic architecture necessary for transgressive segregation is common in both natural and domesticated populations. Philos Trans R Soc Lond B 358: 1141–1147.
Roberts RB, Ser JR, Kocher TD (2009). Sexual conflict resolved by invasion of a novel sex determiner in Lake Malawi cichlid fishes. Science 326: 998–1001.
Saccone G, Peluso I, Artiaco D, Giordano E, Bopp D, Polito LC (1998). The Ceratitis capitata homologue of the Drosophila sex-determining gene Sex-lethal is structurally conserved, but not sex-specifically regulated. Development 125: 1495–1500.
Sanchez L (2008). Sex-determining mechanisms in insects. Int J Dev Biol 52: 837–856.
Seidel HS, Rockman MV, Kruglyak L (2008). Widespread genetic incompatibility in C. elegans maintained by balancing selection. Science 319: 589–594.
Ser JR, Roberts RB, Kocher TD (2010). Multiple interacting loci control sex determination in Lake Malawi cichlid fish. Evolution 64: 486–501.
Shaham S ed. (2006). WormBook: Methods in Cell Biology (January 02, 2006). In: The C. elegans Research Community (ed). WormBook; doi 10.1895/wormbook.1.49.1 http://www.wormbook.org.
Sokol SB, Kuwabara PE (2000). Proteolysis in Caenorhabditis elegans sex determination: cleavage of TRA-2A by TRA-3. Genes Dev 14: 901–906.
Takehana Y, Hamaguchi S, Sakaizumi M (2008). Different origins of ZZ/ZW sex chromosomes in closely related medaka fishes, Oryzias javanicus and O. hubbsi. Chromosome Res 16: 801–811.
Tarone AM, Nasser YM, Nuzhdin SV (2005). Genetic variation for expression of the sex determination pathway genes in Drosophila melanogaster. Genet Res 86: 31–40.
True JR, Haag ES (2001). Developmental system drift and flexibility in evolutionary trajectories. Evolution Dev 3: 109–119.
van Doorn GS, Kirkpatrick M (2007). Turnover of sex chromosomes induced by sexual conflict. Nature 449: 909–912.
Vandeputte M, Dupont-Nivet M, Chavanne H, Chatain B (2007). A polygenic hypothesis for sex determination in the European sea bass Dicentrarchus labrax. Genetics 176: 1049–1057.
Vieira C, Pasyukova EG, Zeng Z-B, Hackett JB, Lyman RF, Mackay TFC (2000). Gentoype-environment interaction for quantitative trait loci affecting life span in Drosophila melanogaster. Genetics 154: 213–227.
Wade MJ, Johnson NA, Jones R, Siguel V, McNaughton M (1997). Genetic variation segregating in natural populations of Tribolium castaneum affecting traits observed in hybrids with T. freemani. Genetics 147: 1235–1247.
Wang S, Basten CJ, Zeng Z-B (2007). Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC http://statgen.ncsu.edu/qtlcart/WQTLCart.htm.
Werren JH, Hatcher MJ, Godfray HCJ (2002). Maternal-offspring conflict leads to the evolution of dominant zygotic sex determination. Heredity 88: 102–111.
Whitfield LS, Lovellbadge R, Goodfellow PN (1993). Rapid sequence evolution of the mammalian sex-determining gene sex-determining gene Sry. Nature 364: 713–715.
Wilkins AS (1995). Moving up the hierarchy: a hypothesis on the evolution of a genetic sex determination pathway. Bioessays 17: 71–77.
Wolff JR, Zarkower D (2008). Somatic sexual differentiation in Caenorhabditis elegans. Curr Top Dev Biol 83: 1–39.
Zauner H, Sommer RJ (2007). Evolution of robustness in the signaling network of Pristionchus vulva development. Proc Natl Acad Sci USA 104: 10086–10091.
Acknowledgements
I thank G Chadderdon, R Den Adel, A Heun, T Mitchell and T Pepper for assistance in the laboratory, and F Janzen, E Myers, I Dworkin, the Janzen laboratory, as well as two anonymous reviewers for very helpful constructive criticism on earlier versions of this paper. A Bronikowski, F Janzen, J A Powell-Coffman, S Proulx, J Serb and J Wendel also provided valuable discussions and advice. J Hodgkin, P Phillips and the Caenorhabditis Genetics Center generously provided worm strains, and photographs of worms for phenotypic analysis were taken at the Iowa State University (ISU) Microscopy and Nano-Imaging Facility. H Teotonio provided helpful guidance in the choice of SNP genotyping methods. This study was funded by the ISU Department of Ecology, Evolution, and Organismal Biology, the ISU Center for Integrated Animal Genomics, the Society for Integrative and Comparative Biology, the Iowa Academy of Sciences and Iowa Science Foundation and an ISU Ecology and Evolutionary Biology Bill Clark Award.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Chandler, C. Cryptic intraspecific variation in sex determination in Caenorhabditis elegans revealed by mutations. Heredity 105, 473–482 (2010). https://doi.org/10.1038/hdy.2010.62
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2010.62
Keywords
This article is cited by
-
Cryptic genetic variation: evolution's hidden substrate
Nature Reviews Genetics (2014)
-
The future of model organisms in human disease research
Nature Reviews Genetics (2011)
-
Hidden variation mapped
Heredity (2010)
