
Abstract
Polyploid plants often produce new phenotypes, exceeding the range of variability existing in the diploid gene pool. Several hundred citrus allotetraploid hybrids have been created by somatic hybridization. These genotypes are interesting models to study the immediate effects of allopolyploidization on the regulation of gene expression. Here, we report genome-wide gene expression analysis in fruit pulp of a Citrus interspecific somatic allotetraploid between C. reticulata cv ‘Willowleaf mandarin’+C. limon cv ‘Eureka lemon’, using a Citrus 20K cDNA microarray. Around 4% transcriptome divergence was observed between the two parental species, and 212 and 160 genes were more highly expressed in C. reticulata and C. limon, respectively. Differential expression of certain genes was confirmed by quantitative real-time RT-PCR. A global downregulation of the allotetraploid hybrid transcriptome was observed, as compared with a theoretical mid parent, for the genes displaying interspecific expression divergence between C. reticulata and C. limon. The genes underexpressed in mandarin, as compared with lemon, were also systematically repressed in the allotetraploid. When genes were overexpressed in C. reticulata compared with C. limon, the distribution of allotetraploid gene expression was far more balanced. Cluster analysis on the basis of gene expression clearly indicated the hybrid was much closer to C. reticulata than to C. limon. These results suggest there is a global dominance of the mandarin transcriptome, in consistence with our previous studies on aromatic compounds and proteomics. Interspecific differentiation of gene expression and non-additive gene regulation involved various biological pathways and different cellular components.
Similar content being viewed by others


Introduction
Polyploidization is a prominent mechanism of speciation in flowering plants and has a strong impact on plant species diversity (Wendel and Doyle, 2005). Polyploid plants often produce new phenotypes, exceeding the range of variability existing in the diploid gene pool (Osborn et al., 2003) and it is generally considered that allopolyploid plants confer an evolutionary advantage over their progenitor (Salmon et al., 2005; Wendel and Doyle, 2005; Chen, 2007). Genomic reorganization, independent evolution and subfunctionalization of duplicated genes (Lynch and Force, 2000; Adams et al., 2003), are important elements of the evolution of polyploid populations. Recent studies have also demonstrated that besides structural changes (Madlung et al., 2005), important modifications of gene expression occur immediately after allopolyploidization events (Comai et al., 2000; Adams et al., 2004; Wang et al., 2004, 2006b). Gene silencing is frequent in allotetraploids. It varies between 1 and 5% in corn allopolyploids (Kashkush et al., 2002) and between 2.5 and 11% in neo-allopolyploids between Arabidopsis thaliana and A. arenosa (Lee and Chen, 2001; Wang et al., 2004), reaching 25% in allotetraploid cotton plants (Adams et al., 2003). Several studies suggest that these variations in expression are repeatable for particular genes. Thus, in Arabidobsis the same set of three genes is affected in four neo-allopolyploid lines arising from independent events of polyploidization (Wang et al., 2004). However, in the same study, gene silencing seemed to be stochastic in other parts of the genome. In cotton, very similar skews have been observed for several recently generated allotetraploid artificial hybrids and natural allotetraploid cotton plants, polyploidization event of which goes back over 1 million years. Moreover, in cotton the neo-regulation of the genome seems to be organ specific (Adams et al., 2003, 2004).
It is generally considered that a ‘genomic shock phenomenon’, as proposed by McClintock (1984), occurs rapidly in interspecific hybrids and allopolyploids, resulting in demethylation of retroelements, relaxation of gene imprinting, and silencing and activation of homoeologous genes, including rRNA genes subjected to nucleolar dominance (Chen, 2007). RNA interference, action of siRNAs and miRNAs and their implication in gene regulation, are also good candidates to explain non-additive gene expression (Chen et al., 2008). Moreover, allopolyploidy induces the juxtaposition and interaction of divergent homeologous regulatory networks, leading to new cis- and trans-acting effects on gene expression (Chen, 2007; Landry et al., 2007) and thus to non-additivity.
In most of these published studies, recent allotetraploid plants arise from sexual crossing with potential intrafamily variability in the case of partial homozygosity of the parental lines. Within this framework, somatic hybrids are interesting models to study the immediate effects of allopolyploidization on the regulation of gene expression. In the last two decades, somatic hybridization has become an important tool for citrus breeding and several hundreds of interspecific and intergeneric allotetraploid hybrids have been synthesized (Grosser et al., 2000). Theoretically, all dominant traits of complementary heterozygous parents can be cumulated in somatic hybrid rootstocks (Grosser et al., 2000). Somatic hybridization has also been widely used to diversify the tetraploid gene pool to be used for triploid citrus breeding (Ollitrault et al., 2008). Allotetraploid citrus from somatic hybridization displays certain transgressive morphological vegetative traits (leaf thickness, stomate density, size and so on) similar to autotetraploids arising from chromosomal doubling of nucellar cells (Ollitrault et al., 2008), which can be associated with tetraploidy per se. However, inheritance of others traits is clearly linked with the parental combinations with codominance or dominance of one or the other parents according to the considered trait. In a study on leaf volatile compounds of seven citrus somatic allotetraploid hybrids sharing Willowleaf mandarin as their common parent (Gancel et al., 2003), including Willowleaf+Eureka lemon hybrid, a systematic dominance of mandarin traits was observed. Particularly noticeable were the absence of monoterpene aldehydes and monoterpene alcohols and the very low levels of sesquiterpene hydrocarbons, sesquiterpene alcohols and sesquiterpene aldehydes (β- and α-sinensals) in all hybrids, as these compounds were found at high concentrations in the non-mandarin parents. Moreover, some specific compounds of Willowleaf mandarin, such as methyl N-methylanthranilate, were found in all hybrids. Such dominance of mandarin traits was also observed under 2-D electrophoresis for the leaf proteome of two allotetraploid somatic hybrids combining Willowleaf mandarin (Citrus reticulata) with Mexican lime (C. aurantifolia) and kumquat (Fortunella margarita). The two allotetraploid hybrids were found to be closer to their mandarin parent than to their other parents in terms of presence/absence of protein spots as well as at a quantitative expression level (Gancel et al., 2006). More recently, Bassene et al. (2009b) selected the allotetraploid C. reticulata cv ‘Willowleaf’+C. limon cv ‘Eureka’ as a model for the analysis of phenotypical inheritance of fruit quality traits in allotetraploid citrus somatic hybrids. Indeed the two parents displayed a very strong differentiation for primary and secondary metabolite profiles as well as for pomological traits. Strong dominance of lemon was found for acid compounds, partial dominance of lemon was found for sucrose and glucose, whereas dominance of mandarin was observed for fructose and most of the aromatic compounds except methyl N-methylanthranilate (Bassene et al., 2009b). Some morphological characteristics of the fruits, such as shape and colour, were closer to mandarin whereas additive inheritance was observed for other traits, such as fruit size and diameter, number of seeds and number of embryos per seed. It is widely accepted that mandarin (C. reticulata), pummelo (C. maxima) and citron (C. medica) are the three basic taxa from which the following cultivated species originated: C. sinensis (sweet orange), C. aurantium (sour orange), C. paradisi (grapefruit) and C. limon (lemon) (Nicolosi et al., 2000). The latter species is probably a direct hybrid between C. aurantium and C. medica (Figure 1; Nicolosi et al., 2000). Considering that C. aurantium comes from hybridization between the C. reticulata and C. maxima (Nicolosi et al., 2000), it should be considered that C. limon is a mosaic of genome regions in heterozygosity between C. medica and C. reticulata and regions in heterozygosity between C. medica and C. maxima. Thus, the specific allelic dosage along the genome of the allotetraploid C. reticulata+C. limon somatic hybrid may be 3 C. reticulata+1 C. medica or 2 C. reticulata+1 C. maxima+1 C. medica.

Phylogenetic origin of the somatic alloteraploid hybrid (WLM+EUR).
The main objective of this study was to analyse whether the new phenotype of this complex three-way tetraploid interspecific hybrid, displaying mixed dominant, codominant and transgressive traits (Gancel et al., 2003; Bassene et al., 2009b), is associated with non-additive expression of the two diploid parental genomes. The study is conducted by hybridization of fruit pulp transcriptome on a genome-wide 20K cDNA microarray, which was developed under the Citrus Functional Genomics Project from a huge clementine expressed sequence tags database (CFGP; http://bioinfo.ibmcp.upv.es/genomics/cfgpDB; Martinez-Godoy et al., 2008).
Materials and methods
Plant material
The study was conducted on fruits of Willowleaf mandarin SRA 133 (C. reticulata Blanco.), Eureka lemon SRA 4 (Citrus limon (L.) Burm.) and their somatic allotetraploid called WLM+EUR (Citrus reticulata Blanco)+(Citrus limon (L.) Burm.). The allotetraploid, WLM+EUR, was produced by symmetric protoplast electrofusion (Ollitrault et al., 2000). This hybrid was genomically characterised by (Bassene et al., 2009a). The 26 polymorphic simple sequence repeat nuclear markers analysed did not reveal any departure from complete addition of the two parental nuclear genomes. Moreover, 36 chromosomes were observed, as expected from addition of the genomes of the two diploid parental species (2 × =18). Chloroplastic simple sequence repeats and mitochondrial sequence-tagged sites revealed that the allotetraploid nuclear genome was combined with chloroplasts and mitochondria of mandarin.
The somatic allotetraploid and the two parents were all grafted onto Volkamer lemon rootstock (Citrus limonia Obs.) and three trees of each genotype were randomly planted in the same field at the Station de Recherche Agronomiques (INRA/CIRAD) in San Giuliano (Corsica, France). From the same experimental field, Bassene et al. (2009a) harvested fruits from three trees at 15-day intervals from December to February 2008 (180–255 days after anthesis (DAA)) and analysed the evolution of juice, sugar and acid contents. The 225 DAA seemed the better date to fit with classical values of maturity indicators for mandarin and lemon. They selected this date for the characterization of aromatic compounds at maturity. The present experiment has been carried out with samples of four fruits per tree harvested at the same date (225 DDA) from three individual trees. At this date, the values of the maturity indicators, respectively, for Willoleaf mandarin, the somatic hybrid and Eureka lemon were: juice content (32±2.3; 26±1.2 and 30±0.8%), soluble solid content (10.1±0.1, 7.1±0.2 and 6.8±0.1%), titratable acidity (0.9±0.1, 3.6±0.1 and 5.3±0.2%) and soluble solid content/titratable acidity ratio (11.2±1.0, 1.97±0.51 and 1.28±0.31). Juice content was expressed as percentage of fruit weight. Titratable acidity of juices was determined by titration to pH 8.2 with 0.1 mol l−1 NaOH and expressed as percentage of anhydrous citric acid, and soluble solid content was determined with a refractometer (Atago model 0–32%, Bellevue, WA, USA). Moreover, fruits displayed mature external colour.
RNA extraction and microarray hybridization
Total RNA was extracted from fruits (225 DAA) according to the protocol described by Ancillo et al. (2007), except that the high pH of the lemon fruit homogenates was neutralized by adding NaOH up to pH-neutral to ensure a better stability of RNA. Extracted RNA was then used in an amplification reaction with the AminoAllyl Message AmpTM aRNA Amplification kit (Ambion, Austin, TX, USA) following the manufacturer's instructions. Then 7.5 μg of UTP-aminoallyl-amplified RNA was labelled using Cy3 or Cy5 dye (GE Healthcare, Chalfont St Giles, UK).
For each genotype, fruits were randomly collected from three different field trees. Three biological replicates for each genotype were generated and compared with a global reference, generated from an equimolar mix of amplified RNAs from each of the nine trees.
A genome-wide 20K cDNA microarray was used, developed under the Citrus Functional Genomics Project (CFGP; http://bioinfo.ibmcp.upv.es/genomics/cfgpDB). The microarray includes 21 081 putative unigenes of citrus (Martinez-Godoy et al., 2008). Microarray hybridization and washing were performed as described by Martinez-Godoy et al. (2008). Afterwards slides were scanned at 532 nm for the Cy3 and 635 nm for the Cy5 dyes, with a GenePix 4000B scanner (Axon Molecular Devices, Sunnyvale, CA, USA), at a 10-nm resolution and 100% laser power. Photomultiplier tube voltages were adjusted to equal the overall signal intensity for each channel, to increase signal-to-noise ratio and to reduce the number of spots with saturated pixels. Spot intensities were quantified using GenePix Pro 6.0 (Axon Molecular Devices). Spots with a net intensity in both channels below twofold the mean background intensity were considered low-signal spots and removed.
Microarray data analysis
Data were global median normalized using GenePix Pro 6.0 (Axon Molecular Devices) so that the median of the median ratios of every valid spot in the slide was equal to 1. Only probes with valid data in all slides were considered for further analysis (9376 spots).
To detect differentially expressed genes, data were analysed with the significance analysis of microarray package (Tusher et al., 2001) using multi-class comparison with a false discovery rate of 4.18% with no fold-change cut-off. The differentially significant expressed genes were clustered in self-organizing maps (Kohonen, 1990) with Euclidean squared similarity metrics using the GEPAS interface (http://gepas.bioinfo.cipf.es/; (Herrero et al., 2003)). The means and s.d. values from each group for each species were normalized to the median of the expression in WLM samples.
Gene lists were further analysed using Blast2GO (Conesa et al., 2005) to find differential distributions of the gene ontology term between statistically differential genes in each self-organizing map and the other genes in the microarray.
Quantitative real-time RT-PCR
Expression of four selected genes was estimated by quantitative real-time RT-PCR using the SYBR Green assay and the LightCycler System (Roche, Mannheim, Germany). Total RNA preparations were cleaned up with the Rneasy Plant Mini Kit (Qiagen, Hilden, Germany), treated with DNase I (RNase-free DNase set; Qiagen) and adjusted to 20 ng RNA per μl using the Quant-iT RiboGreen RNA assay kit (Invitrogen, Paislay, Scotland) following the manufacturer's instructions. The cDNA was synthesized in a 10-μl reaction volume containing 20 ng of DNase-treated RNA, 2.5 pmol each of forward and reverse primers, 1 U of RNase inhibitor (Applied Biosystems, Foster city, CA, USA), 2 μl of LC FastStart DNA MasterPLUS SYBR Green I (Roche), and 2.5 U of MuLV Reverse Transcriptase (Applied Biosystems). Primer pairs for each gene were designed on the basis of corresponding sequences available in the database of the CFGP (http://bioinfo.ibmcp.upv.es/genomics/cfgpDB) and are shown in Table 1.
The thermal profile consisted of 48 °C for 30 min, 95 °C for 10 min, and 45 cycles of 95 °C for 2 s, 60 °C for 10 s and 72 °C for 15 s followed by a melting programme of 60 °C for 60 s. To transform fluorescent intensity measurements into relative mRNA levels, a 10-fold dilution series of an RNA sample was used as the standard curve. All the experiments were performed in triplicate and means were calculated.
Results
A cDNA microarray containing 21 081 putative unigenes was used to analyse the gene expression profile of the Willowleaf+Eureka lemon allotetraploid hybrid and its two diploid parental genomes. Total RNA was extracted from fruit pulp (225 DAA), amplified, labelled and hybridized to the microarray. Three biological replicates were analysed per species using a global reference experimental design. Microarray data have been archived in NCBI public database (GSE18386; http://www.ncbi.nlm.nih.gov/projects/geo/query/acc.cgi?acc=GSE18386). After quality analysis, 9376 spots (unigenes) were considered as valid for further analyses. The average signal of the three genotypes were not significantly different: respectively, 1.033±0.021, 1.101±0.066 and 1.058±0.041 for the mandarin, the lemon and the hybrid. This suggests that the chip is not biased toward mandarin or lemon. This is a logical result because this chip is based on large fragments (75% are bigger than 400 nucleotides; Martinez-Godoy et al., 2008) and gene homology is very high among cultivated Citrus species. Expression levels were analysed in a multi-class response using significance analysis of microarray (Tusher et al., 2001). In total, 372 differentially expressed genes between the three analysed genotypes were statistically significant, with a false discovery rate of 4.18% meaning that among these 372 genes, 15 should be false positive (Pawitan et al., 2005). Respectively, 100% and more than 77% of the genes classed as differentially expressed, differed in expression by more than 1.5 × and 2 × .
Differential expression of some genes was confirmed by quantitative real-time RT-PCR using the same RNA preparations analysed in hybridization experiments. The relative accumulation of the four mRNAs (alcohol dehydrogenase, phosphoenolpyruvate carboxylase kinase 2, putative sugar transporter and sucrose phosphatase) paralleled that observed by microarray hybridization, although the changes detected by both procedures were not quantitatively identical (Table 1). This variation is commonly observed in validation of microarray results by RT-PCR (Allen and Nuss, 2004; Lopez et al., 2005) and is probably due to intrinsic differences between both techniques.
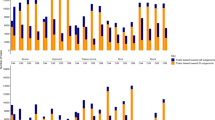
Statistically significant differentially expressed genes were grouped according to self-organizing maps (Kohonen, 1990). To reflect biological differences in gene expression, data were normalized to the median expression of Willowleaf samples. Two global patterns of gene expression were found among these genes. One corresponding to those genes repressed in Eureka lemon with respect to those of mandarin, and a second corresponding to those overexpressed in Eureka lemon compared with Willowleaf mandarin. Group 1 included 212 genes repressed in Eureka lemon relative to mandarin and group 2 included 160 genes overexpressed in Eureka lemon compared with Willowleaf mandarin. To compare the global expression level of these groups of genes in the two parents and the hybrid, means of the expression data (logarithmic data) from each group for each species were normalized to the median of the expression in Willowleaf Mandarin samples. Expression levels in the hybrid were between the two parents (Figure 2) for the two groups, however, they were much closer to those of Willowleaf mandarin for group 2 (Figure 2b).

Comparison of global genes expression level between parents and the allotetraploid. Data are means of expression log data from each species normalized to the median of the expression in Willowleaf mandarin samples. (a) Genes from group 1, (b) Genes from group 2. EUR, Eureka lemon; WLM, Willowleaf mandarin; WLM+EUR, allotetraploid hybrid.
Five genes in group 1 had significant transgressive expression (over mandarin expression) of the somatic hybrid (Table 2), whereas no transgressive expression was observed for group 2. Among the five significantly transgressive genes, two have unknown function. Interestingly, the three other ones are associated with cellular signal transduction in relation with Ca2+.
The distribution of gene expression in the somatic hybrid compared with that of its two parents was analysed in greater depth to analyse the additivity of gene expression. For each group, initial expression data (without logarithmic transformation) were normalised to attribute a null value of gene expression for the parent with low expression and 1 to the parent with high expression. For group 1 (overexpression of mandarin), gene expressions of the allotetraploid are distributed between the two parents (Figure 3a) and taking into account the transgressive genes, the average expression value is 0.70. For group 2 (overexpression of lemon), global non-additivity is observed (Figure 3b) with a strong dominance of mandarin (low expression). Indeed, no gene expression was found over 0.55 and average gene expression is 0.09.

Allotetraploid hybrid gene expression distributed between the two parents and a mid-parent value. Expression data were normalised to attribute a null value of gene expression to the parent with low expression and 1 to the parent with high expression. A theoretical mid-parent value was assigned at 0.5. (a) Genes from group 1, (b) Genes from group 2. L, lemon; LD, lemon dominance; M, mandarin; MD: mandarin dominance; MP, mid-parent; H, hybrid mean value ((a): H=0.70±0.08; (b): H=0.09±0.01). A total of 34 genes presented transgressive over expression.
When all the genes displaying significant differentiation among the three genotypes are considered, the transcriptome of the allotetraploid hybrid is much closer to its mandarin parent than to its lemon parent. This is clearly shown by the cluster analysis (UPGMA: unweighted pair group method with arithmetic mean) based on Euclidian distances calculated from the relative expression of the 372 genes (Figure 4) under consideration.

Cluster analysis (UPGMA: unweighted pair group method with arithmetic mean) based in Euclidian distances calculated from the relative expression of genes displaying significant differentiation among the three genotypes (Willowleaf mandarin; WLM+EUR, allotetraploid hybrid; Eureka lemon).
To examine whether any of the gene ontology terms obtained in the gene groups identified by self-organizing maps were enriched, when compared with the whole microarray gene set taken as a reference, we applied the Fisher's test implemented in Blast2GO (Conesa et al., 2005). The genes are represented in various functional categories. In group 1, the genes attributed to the functional category ‘secretory pathway’ (about 27%) are among the most highly represented, followed by the genes from the category ‘metabolic process’, about 10% (Figure 5a). The rest of the genes are distributed among different biological processes and represent about 7–2%. The category ‘secretory pathway’ designates the process by which proteins are allocated to different destinations in the cell. No significant enrichment was found in this group compared with the reference distribution. Half of the genes from group 1 encoded for proteins located in ‘plastid’ (18%), ‘mitochondrion’ (18%) and ‘cytoplasmic membrane-bounded vesicle’ (13%), but most of the cellular compartments are represented (Figure 5b). The genes in the second group are also highly represented (27%) in the category ‘secretory pathway’, whereas the other categories are represented equally (Figure 6a). In this group, genes related with ‘secondary metabolic processes’, ‘photosynthesis’ and ‘photosystems I and II’ were found significantly enriched as compared with the reference distribution (Table 3). The location of the proteins encoded by these genes is very similar to that in group 1 (Figure 6b).

Classification of genes from group 1 among gene ontology (GO) functional categories. (a) GO Biological Process categories. (b) GO Cellular Component categories.

Classification of genes from group 2 among gene ontology (GO) functional categories. (a) GO Biological Process categories. (b) GO Cellular Component categories.
Discussion
Owing to the low availability of field samples, the experiment was carried out with three replicates, which led to high data variability in that the number of significant genes obtained was probably underestimated. Nevertheless, among the 9376 spots (unigenes) considered as valid, 372 genes (4%) displayed significant differential gene expression among the three genotypes. After these genes were normalized to the average expression of Willowleaf samples, they automatically clustered into two groups, namely those which are overexpressed in mandarin vs lemon (group 1) and those which are overexpressed in lemon vs mandarin (group 2). So it seems we have not identified any genes with significant transgressive expression for the allotetraploid hybrid without gene expression differentiation between mandarin and lemon.
Non-additivity is frequent among the genes displaying significant interspecific differences in expression and leads to global mandarin transcriptome dominance
In this study of the transcriptome of citrus-fruit pulp, we detected 4% divergence between the two parental species. A total of 212 and 160 genes were highly expressed in C. reticulata and C. limon, respectively. This is below the 15% observed by Wang et al. (2006b) between Arabidopsis thaliana and A. arenosa in a microarray study of leaf transcriptome or the 10% divergence found between maize lines (Swanson-Wagner et al., 2006).
According to the additivity hypothesis, newly synthesized allopolyploids are supposed to display mid-parent expression patterns. Using a microarray without allelic specificity, we cannot detect the situation in which repression of an allele is compensated by increased expression of its homologous allele. Thus, we should bear in mind that microarray analysis may underestimate the number of genes displaying allelic differential expression between the allotetraploid line and the parents.
The genes displaying interspecific expression divergence between mandarin and lemon displayed global non-additive behaviour in the WLM+EUR allotetraploid hybrid. Indeed, after linear normalization, giving a value of 1 for the overexpressed parental genotype and 0 for the underexpressed one, we observed that most of the genes with underexpression for mandarin were subjected to repression in the allotetraploid (mid value for the hybrid=0.09). When mandarin was overexpressed compared with lemon, the gene distribution was much more balanced. It should also be noted that transgressive overexpression was only observed when mandarin was overexpressed compared with lemon.
A global trend for downregulation was observed by Wang et al. (2006b) in a study of neo-allotetraploid lines between Arabidopsis arenosa and A. thaliana. Indeed, among the genes with non-additive expression, they observed that 76 and 65% were, respectively, downregulated in two independent allotetraploid genotypes. They suggested that repression is a mode of non-additive gene regulation in synthetic allotetraploids. Moreover, as for the WLM+EUR allotetraploid, they observed that most of the genes with non-additive expression were part of those displaying significant differentiation of parental expression. Downregulation of interspecific hybrid genes was also observed for sunflower (Lai et al., 2006). However, at intraspecific level in maize, Swanson-Wagner et al. (2006) observed that only 22% of the differentially regulated genes exhibited non-additive modes of gene expression. Moreover, 181 of these 305 non-additive genes exhibited high-parental dominance and only 23 exhibited low-parental dominance. More systematic non-additive behaviour of the gene with differential parental expression and downregulation must be associated with interspecificity and global genetic divergences of parental genomes, leading to regulation incompatibilities (Landry et al., 2007). In addition to the aforementioned species, deviations to additivity were found in synthetic allopolyploids of Senecio (Hegarty et al., 2006), Brassica (Albertin et al., 2006), Triticum (He et al., 2003) and Gossypium neo-polyploids (Flagel et al., 2008), suggesting that the differential regulation of gene expression is a common feature of plant allopolyploids.
We found a strong global dominance of the mandarin transcriptome on considering all the genes with significant differences between the two parental genotypes. Such transcriptome dominance has been demonstrated in Arabidopsis allotetraploid between A. arenosa and A. thaliana. Indeed Wang et al. (2006b) observed that the genes more highly expressed in A. thaliana autotetraploids than in A. arenosa are subject to orchestrated repression in the synthetic allotetraploids. Indeed, considering the mid-value of the two allotetraploid lines analysed, 97% of the genes overexpressed in A. thaliana were repressed whereas such under expression was only observed for 47% of the genes overexpressed in A. arenosa. They related this result with the silencing of A. thaliana rRNA genes subjected to nucleolar dominance and with overall suppression of the A. thaliana phenotype in the synthetic allotetraploids and natural A. suecica, a putative old natural hybrid between A. arenosa and A. thaliana. Transcriptome dominance has also been reported in two recent studies on cotton (Gossypium hirsutum L.) and demonstrates that transcriptome dominance should tend towards one or the other parent depending on the organ analysed. Upland cotton was formed by ancient interspecific hybridization between AA and DD genome species. In a large microarray study of ovular tissue, Yang et al. (2006) demonstrated that AA subgenome genes of all functional classifications were selectively enriched in G. hirsutum L., whereas in a recent study of petal tissues, Flagel et al. (2008) encountered the opposite generalized bias and concluded that neither Gossypium genome is globally dominant with respect to expression, but rather, each genome may have local dominance in certain tissue types or developmental stages. Therefore, transcriptome dominance is probably a general consequence of hybridization effects on gene expression in interspecific hybrids and allopolyploids, and seems to be potentially oriented differently in different organs as a probable result of subfunctionalization of duplicated genes (Lynch and Force, 2000).
Interspecific differential gene regulation and non-additivity in WLM+EUR allotetraploid are poorly related to functional categories or cellular component localization of gene products
One of the important benefits of microarray studies is that the expression of thousands of genes can be analysed simultaneously. Thus, inferences can be drawn about the types and categories of genes, expression of which is affected by allopolyploidy. We observed that, for the genes overexpressed for mandarin compared with lemon, no particular functional category of genes was overly affected between lemon and mandarin. Regarding the reciprocal situation, significant enrichment was found for the categories ‘secondary metabolic processes’, ‘photosynthesis’ and ‘photosystems I and II’; however, they only represent 25 of the 160 genes in this group. No specific enrichment of cellular component was found for the genes with differential expression, which means that most of the cellular components are involved. A microarray study of Senecio allopolyploid and hybrid species showed that the expression of genes with no particular functional category was overly affected, although genes involved in floral or pollen development were slightly over-represented (Hegarty et al., 2006). Similarly, a microarray study of the homoploid hybrid species Helianthus deserticola showed differential regulation of genes in various functional categories in the hybrid (Lai et al., 2006). A microarray study of Arabidopsis neo-polyploids also showed differential expression of genes in a variety of functional categories (Wang et al., 2006b). However, in the latter study, genes involved in hormonal regulation were highly represented, particularly the ethylene biosynthesis pathway and cell defence mechanisms. Overall, it seems that expression of a variety of types and categories of genes, including gene products in all cellular components, is altered in allopolyploids and hybrids, but certain species or organ types may show biases for certain types of genes.
Non-additive gene expression may lead to dominance or new phenotype in allopolyploids
Non-additive expression in allopolyploids can affect phenotypic traits. The global dominance of mandarin transcriptome in the WLM+EUR allotetraploid hybrid is coherent with the results of Gancel et al. (2006), who described a proteome dominance of mandarin cv ‘Willowleaf’ when combined by somatic hybridization with Mexican lime (C. aurantifolia) and Kumquat cv ‘Marumi’ (F. japonica). These results suggest a generalized behaviour of dominance of the mandarin both at transcriptomic and proteomic level. Gancel et al. (2006) linked this proteome dominance to the dominance of the leaf aromatic pattern of mandarin over the ones of all the other parental species combined by somatic hybridization (Gancel et al., 2003). It was also demonstrated in Brassica neo-polyploids that protein abundance can be altered by allopolyploidy (Albertin et al., 2006). A relatively high number of proteins displayed quantitative differences in polyploids as compared with their parents, but few proteins appeared or disappeared.
In a recent study, Bassene et al. (2009b) described the relationship between the neo-expression of the genes of the carotenoid pathway and the new pattern of carotenoid composition of juice sac in the allotetraploid hybrid WLM+EUR. Indeed, underexpression of the first step in the pathway and transgressive overexpression of the last step, producing abcissic acid from violaxanthin, lead to very low levels of total carotenoid but with all the compounds found in mandarin. Another example is a recent study of genes involved in regulating flowering time: flowering locus C (FLC; negatively regulates flowering) and FRI (positively regulates FLC). Meanwhile, in neo-polyploid Arabidopsis, expression levels of FLC derived from the A. thaliana parent in the allopolyploid are reported to be considerably upregulated, whereas expression levels of FLC from the A. arenosa parent are downregulated (Wang et al., 2006a). As a result, flowering time in neo-polyploids was later than that in either parent. However, the nucleolar dominance of A. arenosa over A. thaliana (Pikaard, 1999) is associated with the overall suppression of A. thaliana phenotype in new allotetraploids and in natural A. suecica (Wang et al., 2006b). Results regarding H. deserticola suggest that non-additive gene regulation and particularly transgressive expression of transport-related proteins should be related with its high adaptability to water-stress conditions (Lai et al., 2006).
Our results for the WLM+EUR allotetraploid show that gene underexpression of mandarin compared with its other parent is highly dominant; however, when mandarin genes are overexpressed the allotetraploid can display, additivity, lemon or mandarin dominance as well as transgressive (over) gene expression. It is logical to hypothesize that the phenotype of mandarin will be generally dominant when phenotypic differentiation between mandarin and lemon is associated with underexpression of the mandarin genes. Similarly, in the opposite situation (overexpression of mandarin genes) there should be codominance, phenotypic mandarin or lemon dominance as well as overdominance. Interestingly, the group of genes with underexpression of mandarin and generalized dominance of mandarin transcriptome displays significant enrichment for the functional category ‘secondary metabolite processes’.
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
Adams KL, Cronn R, Percifield R, Wendel JF (2003). Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc Natl Acad Sci USA 100: 4649–4654.
Adams KL, Percifield R, Wendel JF (2004). Organ-specific silencing of duplicated genes in a newly synthesized cotton allotetraploid. Genetics 168: 2217–2226.
Albertin W, Balliau T, Brabant P, Chevre AM, Eber F, Malosse C et al. (2006). Numerous and rapid nonstochastic modifications of gene products in newly synthesized Brassica napus allotetraploids. Genetics 173: 1101–1113.
Allen TD, Nuss DL (2004). Specific and common alterations in host gene transcript accumulation following infection of the chestnut blight fungus by mild and severe hypoviruses. J Virol 78: 4145–4155.
Ancillo G, Gadea J, Forment J, Guerri J, Navarro L (2007). Class prediction of closely related plant varieties using gene expression profiling. J Exp Bot 58: 1927–1933.
Bassene JB, Berti L, Costantino G, Carcouet E, Kamiri M, Tomi F et al. (2009a). Inheritance of characters involved in fruit quality in a citrus interspecific allotetraploid somatic hybrid. J Agric Food Chem 57: 5065–5070.
Bassene JB, Froelicher Y, Dhuique-Mayer C, Mouhaya W, Ancillo G, Morillon R et al. (2009b). Non additive phenotypic and transcriptomic patterns of the carotenoid biosynthesis pathway in the pulp of a citrus allotetraploid somatic hybrid. Plant Cell Report 28: 1689–1697.
Chen M, Ha M, Lackey E, Wang J, Chen ZJ (2008). RNAi of met1 reduces DNA methylation and induces genome-specific changes in gene expression and centromeric small RNA accumulation in Arabidopsis allopolyploids. Genetics 178: 1845–1858.
Chen ZJ (2007). Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu Rev Plant Biol 58: 377–406.
Comai L, Tyagi AP, Winter K, Holmes-Davis R, Reynolds SH, Stevens Y et al. (2000). Phenotypic instability and rapid gene silencing in newly formed Arabidopsis allotetraploids. Plant Cell 12: 1551–1567.
Conesa A, Gotz S, García-Gómez JM, Terol J, Talón M, Robles M (2005). Blast2go: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676.
Flagel L, Udall J, Nettleton D, Wendel J (2008). Duplicate gene expression in allopolyploid Gossypium reveals two temporally distinct phases of expression evolution. BMC Biol 6: 16.
Gancel AL, Grimplet J, Sauvage FX, Ollitrault P, Brillouet JM (2006). Predominant expression of diploid mandarin leaf proteome in two citrus mandarin-derived somatic allotetraploid hybrids. J Agric Food Chem 54: 6212–6218.
Gancel AL, Ollitrault P, Froelicher Y, Tomi F, Jacquemond C, Luro F et al. (2003). Leaf volatile compounds of seven citrus somatic tetraploid hybrids sharing Willowleaf mandarin (Citrus deliciosa Ten) as their common parent. J Agric Food Chem 51: 6006–6013.
Grosser J, Ollitrault P, Olivares O (2000). Somatic hybridization in Citrus: an effective tool to facilitate variety improvement. In Vitro Cell Dev Biol Plant 36: 434–449.
He P, Friebe BR, Gill BS, Zhou JM (2003). Allopolyploidy alters gene expression in the highly stable hexaploid wheat. Plant Mol Biol 52: 401–414.
Hegarty MJ, Barker GL, Wilson ID, Abbott RJ, Edwards KJ, Hiscock SJ (2006). Transcriptome shock after interspecific hybridization in Senecio is ameliorated by genome duplication. Curr Biol 16: 1652–1659.
Herrero J, Al-Shahrour F, Diaz-Uriarte R, Mateos A, Vaquerizas JM, Santoyo J et al. (2003). GEPAS: a web-based resource for microarray gene expression data analysis. Nucleic Acids Res 31: 3461–3467.
Kashkush K, Feldman M, Levy AA (2002). Gene loss, silencing and activation in a newly synthesized wheat allotetraploid. Genetics 160: 1651–1659.
Kohonen T (1990). The self-organizing map. Proc IEEE 78: 1464–1480.
Lai Z, Gross BL, Zou Y, Andrews J, Rieseberg LH (2006). Microarray analysis reveals differential gene expression in hybrid sunflower species. Mol Ecol 15: 1213–1227.
Landry CR, Hartl DL, Ranz JM (2007). Genome clashes in hybrids: insights from gene expression. Heredity 99: 483–493.
Lee HS, Chen ZJ (2001). Protein-coding genes are epigenetically regulated in Arabidopsis polyploids. Proc Natl Acad Sci USA 98: 6753–6758.
Lopez C, Soto M, Restrepo S, Piegu B, Cooke R, Delseny M et al. (2005). Gene expression profile in response to Xanthomonas axonopodis pv. manihotis infection in cassava using a cDNA microarray. Plant Mol Biol 57: 393–410.
Lynch M, Force AG (2000). The probability of duplicate gene preservation by subfunctionalization. Genetics 154: 459–473.
Madlung A, Tyagi AP, Watson B, Jiang HM, Kagochi T, Doerge RW et al. (2005). Genomic changes in synthetic Arabidopsis polyploids. Plant J 41: 221–230.
Martinez-Godoy MA, Mauri N, Juarez J, Marques MC, Santiago J, Forment J et al. (2008). A genome-wide 20K citrus microarray for gene expression analysis. BMC Genomics 9: 318.
McClintock B (1984). The significance of responses of the genome to challenge. Science 226: 792–801.
Nicolosi E, Deng ZN, Gentile A, Malfa Sl, Continella G, Tribulato E (2000). Citrus phylogeny and genetic origin of important species as investigated by molecular markers. Theor Appl Genet 100: 1155–1166.
Ollitrault P, Dambier D, Froelicher Y, Carreel F, d’Hont A, Luro F et al. (2000). Somatic hybridisation potential for citrus germplasm utilization. Cah Agric 9: 223–236.
Ollitrault P, Dambier D, Luro F, Froelicher Y (2008). Ploidy manipulation for breeding seedless triploid citrus. Plant Breed Rev 30: 323–352.
Osborn TC, Pires JC, Birchler JA, Auger DL, Chen ZJ, Lee HS et al. (2003). Understanding mechanisms of novel gene expression in polyploids. Trends Genet 19: 141–147.
Pawitan Y, Michiels S, Koscielny S, Gusnanto A, Ploner A (2005). False discovery rate, sensitivity and sample size for microarray studies. Bioinformatics 21: 3017–3024.
Pikaard CS (1999). Nucleolar dominance and silencing of transcription. Trends Plant Sci 4: 1360–1385.
Salmon A, Ainouche ML, Wendel JF (2005). Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Mol Ecol 14: 1163–1175.
Swanson-Wagner RA, Jia Y, Decook R, Borsuk LA, Nettleton D, Schnable PS (2006). All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc Natl Acad Sci USA 103: 6805–6810.
Tusher VG, Tibshirani R, Chu G (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121.
Wang J, Tian L, Madlung A, Lee HS, Chen M, Lee JJ et al. (2004). Stochastic and epigenetic changes of gene expression in Arabidopsis polyploids. Genetics 167: 1961–1973.
Wang JL, Tian L, Lee HS, Chen ZJ (2006a). Nonadditive regulation of FRI and FLC loci mediates flowering-time variation in Arabidopsis allopolyploids. Genetics 173: 965–974.
Wang JL, Tian L, Lee HS, Wei NE, Jiang HM, Watson B et al. (2006b). Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 172: 507–517.
Wendel J, Doyle J (2005). Polyploidy and evolution in plants. In: Henry RJ (ed). Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants. CABI: Oxfordshire, UK. pp 97–117.
Yang SS, Cheung F, Lee JJ, Ha MS, Wei NE, Sze SH et al. (2006). Accumulation of genome-specific transcripts, transcription factors and phytohormonal regulators during early stages of fiber cell development in allotetraploid cotton. Plant J 47: 761–775.
Acknowledgements
This study was subsidised by the Collectivité Territoriale de Corse, the ‘Triploid mandarin breeding project (Proyectos de Investigación Fundamental no orientada; AGL 2008-00596) and the ‘Biotechnologia de citricos’ project (Prometeo 2008/12).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Bassene, J., Froelicher, Y., Dubois, C. et al. Non-additive gene regulation in a citrus allotetraploid somatic hybrid between C. reticulata Blanco and C. limon (L.) Burm. Heredity 105, 299–308 (2010). https://doi.org/10.1038/hdy.2009.162
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2009.162
Keywords
This article is cited by
-
Comprehensive mechanistic insights into the citrus genetics, breeding challenges, biotechnological implications, and omics-based interventions
Tree Genetics & Genomes (2022)
-
Identification of differentially expressed transcripts at critical developmental stages in sorghum [Sorghum bicolor (L.) Moench] in relation to grain yield heterosis
3 Biotech (2019)
-
Gene Expression Analysis in Sorghum Hybrids and Their Parental Lines at Critical Developmental Stages in Relation to Grain Yield Heterosis by Exploiting Heterosis-Related Genes from Major Cereals
Plant Molecular Biology Reporter (2018)
-
Comparative metabolic and transcriptional analysis of a doubled diploid and its diploid citrus rootstock (C. junos cv. Ziyang xiangcheng) suggests its potential value for stress resistance improvement
BMC Plant Biology (2015)
-
An RNA-seq transcriptome analysis of floral buds of an interspecific Brassica hybrid between B. carinata and B. napus
Plant Reproduction (2014)
