
Abstract
The success of regeneration attempt is based on an ideal combination of stem cells, scaffolding and growth factors. Tissue constructs help to maintain stem cells in a required area for a desired time. There is a need for easily obtainable cells, potentially autologous stem cells and a biologically acceptable scaffold for use in humans in different difficult situations. This study aims to address these issues utilizing a unique combination of stem cells from gingiva and a hydrogel scaffold, based on a natural product for regenerative application. Human gingival mesenchymal stem cells (HGMSCs) were, with due induction, differentiated to neuronal lineages to overcome the problems associated with birth tissue-related stem cells. The differentiation potential of neuronal lineages was confirmed with suitable specific markers. The properties of mesenchymal stem cells in encapsulated form were observed to be similar to free cells. The encapsulated cells (3D) were then subjected to differentiation into neuronal lineages with suitable inducers, and the morphology and gene expression of transient cells were analyzed. HGMSCs was differentiated into neuronal lineages as both free and encapsulated forms without any significant differences. The presence of Nissl bodies and the neurite outgrowth confirm the differentiation. The advantages of this new combination appear to make it a promising tissue construct for translational application.
Similar content being viewed by others

Introduction
Despite its promises and the huge investments in it, stem cell therapy is far from being utilized to its full potential. Although it has been employed in many regenerative procedures, its maximum use has not been exploited. Although this lack of maximum usage can be attributed to various reasons, an important factor is the ideal coexistence of cells, scaffolds and signals. Combination and permeation have augmented its use and success in a few situations but not all. It is always desirable to have stem cells that are easy to procure with minimal morbidity and invasiveness to the host and do not initiate an immune reaction. The cells obtained must be pluripotent to generate tissue and to have positive markers of self-renewal and differentiation (e.g., Oct-4/Nanog). It is even more desirable if the procedure to procure the cells is simple and if the cells can be obtained from both sexes.
The mesenchymal stem cells (MSCs) of birth-associated tissue with pluripotency have been accepted as nature’s gift, but the accessibility and availability are cumbersome. Although dental pulp is highly potential, the removal of this tissue leads to non-vitality. Gingiva, one of the tissues bestowed with a high regenerative capacity, could be the best source of MSCs.1 Its origin is neural crest, and the differentiation to different lineages supports the use of gingival tissue cells for regeneration. In addition, the reported positive results2 on mesenchymal markers and pluripotency suggest the need for in-depth experimental research on the differentiation of gingival MSCs.
Scaffolds, a three-dimensional (3D) matrix, play an important role in the construction of tissues. The nature of the material used in the preparation, that is, its shape, size, pore size, and physical and mechanical properties,3 decides the fate of the cells. It is worthwhile to use resorbable scaffolds to avoid the drawbacks of a second intervention for scaffold removal. Thus, hydrogels came into the limelight and have been considered a user-friendly scaffold for cell regeneration. Hydrogels of proteins, carbohydrates and polymers of both natural and synthetic origins have been studied extensively for varied applications. The role of hydrogels in cell differentiation and maintenance has been initiated in recent years and is of great benefit to tissue engineering. Recently, Cai et al.4 reported in detail the retention of human adipose stem cells in poly-ethylene glycol-engineered recombinant protein hydrogel and its differentiation. This hydrogel was biocompatible and allowed the cells to retain and differentiate, but the complexity in the preparation and the availability were found to be the major drawbacks.
Neuronal cells fall under the permanent cells category because of the deficiency of progenitors for brain and spinal neuronal cells. For these reasons, many neuronal disorders were claimed to have a poor prognosis. However, with the advent of stem cells, this myth of limited progeny has been put in check. Neuronal lineages of stem cells are beneficial not only for Alzheimer’s and Parkinsonism but also for situations such as spinal cord injuries, where intervention is essential.
Scant information is available on the differentiation of gingival MSCs to neuronal lineages. We aimed to develop a tissue-engineered construct with human gingival mesenchymal stem cells (HGMSCs) due to its known developmental advantages with a biocompatible scaffold that could be utilized directly for translational purposes. Furthermore, the study has been extended to understand the differentiation profile under free (2D) and encapsulated (3D) forms, which will give the most valuable input regarding the vitality of cells to proceed for therapeutic applications. Encapsulation studies were initiated with bioconjugated hydrogel based on its appreciable physical, functional and mechanical properties.5 Thus, the present study was undertaken to explore the neuronal differentiation of gingival MSCs in free form and in encapsulated form in the chosen hydrogel and to authenticate it using various staining procedures, selective markers and gene-expression analysis. The results of the study pave a new way in the field of hydrogel-based stem cell therapy using an easily accessible gingival tissue to meet the challenges of neuronal disorders.
Materials and methods
Tissue source
Ethical clearance was obtained from Institutional Ethics and Stem Cell Ethics Committees of Sri Ramachandra University, Chennai. Human gingival samples were obtained from the Department of Periodontology in the Faculty of Dental Sciences at Sri Ramachandra University. Informed consent was obtained from all five patients, who were in the age group of 20–30 years.
Details on chemicals, reagents and instruments used in the present study
Alpha minimum essential medium (αMEM) and trypsin were purchased from Lonza (Basel, Switzerland).MSC-certified fetal bovine serum and antibiotics were procured from Gibco (Waltham, MA, USA). Collagenase Type II HiChondroXL Chondrogenic differentiation media were obtained from HiMedia (Mumbai, India). All plastic ware was purchased from Nunc Nalgene (Rochester, NY, USA). Stem cell isolation and analysis were performed using a BD Stem Flow kit purchased from BD Pharmingen (Franklin Lakes, NJ, USA). A StemLight Pluripotency Antibody Kit (Cell Signaling Technology, Danvers, MA, USA) was used for the pluripotent analysis. The panel of antibodies β III tubulin and mouse anti-human GFAP were purchased from BioLegend (San Diego, CA, USA). Mouse anti-human MAP-2 was obtained from eBiosciences (San Diego, CA, USA). Anti-S-100 was procured from Biogenex (Fermont, CA, USA). Anti-CD44 was obtained from BD Pharmingen. Secondary antibodies, anti-mouse-IgG fluorochrome-labeled antibody and anti-mouse IgG were obtained from R&D Systems (Minneapolis, MN, USA). Goat anti-mouse IgG-fluorescein isothiocyanate conjugate was obtained from Genei (Bangalore, India). Anti-mouse IgM TRITC was obtained from Sigma-Aldrich (St Louis, MO, USA). Primers for gene expression were purchased from Genei. All other chemicals of an analytical (research) grade were purchased from Sigma Aldrich.
A Nikon TE2000 Eclipse inverted fluorescence microscope with a CCD camera and ImagePro Software and a Nikon Eclipse 80i upright microscope with a camera were employed for observations, imaging and analysis. A Zeiss LSM 510 confocal microscope (Carl Zeiss, Oberkochen, Germany) was used for confocal imaging studies. Flow cytometry analysis was conducted using a BD FACSCalibur (BD Biosciences, Mississauga, ON, Canada). 3H-Thymidine counting was performed using a Geiger Muller counter (Prescott, AZ, USA). A semiquantitative PCR reaction was carried out using an Eppendorf Veriti 96-Well Thermal Cycler (Carlsbad, CA, USA).
Isolation and culturing of HGMSCs
Gingival tissues were obtained during a crown-lengthening procedure or an operculectomy in patients in the age group of 20–30 years. The sampling site, free from clinical signs of inflammation, such as color contour and bleeding, was exposed to 2% lidocaine (1:80 000 adrenalines, local anesthesia). With a scalpel, the gingival margin and the adjacent papilla were excised. The tissue was rinsed with saline. The loose fibrin tags were removed with sharp scissors and then immediately placed in a transport medium (phosphate-buffered saline) with antibiotics. Then, the culturing process proceeded.
The obtained gingival tissues were aseptically cut into 1–3 mm2 pieces and digested at 37 °C for 2 h in a sterile medium containing 1 mg ml−l collagenase type II with gentle agitation. The suspension was filtered through a 70-μm Falcon strainer, and the cell suspension was subjected to centrifugation at 1000 g for 5 min at 37 °C. The cell pellet obtained was resuspended in complete media and used for the present study.6 Gingival cells were distributed evenly into a T75-cm2 flask in complete αMEM supplemented with 10% fetal bovine serum, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 100 μg ml−1 amphotericin B, and 2 mM L-glutamine and cultured at 37 °C with 5% CO2 in a humidified tissue culture incubator. The growth medium was changed every third day. The plastic-adherent confluent cells were passaged with 0.05% trypsin containing 1 mM EDTA, and the cells of the second to sixth passages were used for experiments.
Preliminary Characterization studies on HGMSCs (2D)
Proliferation analysis—3H-thymidine assay
Followed by culturing, HGMSCs were labeled with 3H7 at a final concentration of 1 μCi ml−1 3H-thymidine and incubated for 72 h. After the scheduled time interval, cultures were rinsed three times with ice-cold αMEM and incubated with 1 ml of ice-cold 5% trichloroacetic acid for at least 1 h. They were then centrifuged at 12 000 r.p.m. for 5 min. Then, they were again treated with trichloroacetic acid at the same concentration and then centrifuged. The pellet thus obtained was rinsed with phosphate-buffered saline, transferred into an aluminum planchet and subjected to evaporation under an infrared lamp until a thin film of residue appeared on the planchet. The β activity was counted after 10 min, and the c.p.m. (counts per minute) was calculated accordingly.
Phenotypic marker analysis
Phenotypic characterization was carried out according to ISCT guidelines. In brief, approximately 6 × 106 HGMSCs were incubated with a Human MSC Analysis kit (BD) containing preconjugated and pretitrated cocktails with defined positive and negative expression markers along with the corresponding isotype controls.8 It was subjected to analysis using a BD FACSCalibur flow cytometer.
In vitro differentiation studies on HGMSCs to adipocytes, osteocytes and chondrocytes
Initial attempts were made to assess the differentiation of HGMSCs to adipocytes9 and osteocytes,9 as previously described. Chondrocyte differentiation was carried out with HiChondroXL Chondrogenic Differentiation Medium, as per the manufacturer’s instructions.10
Multipotent marker analysis by immunofluorescence
HGMSCs were cultured in 96-well plates, fixed with ice-cold methanol for 4 min and then treated with 3.7% formaldehyde for 10 min. Sodium borohydride was added as described7 to reduce the autoflourescence, and the cells were blocked with buffer containing 3% normal goat serum and 1% bovine serum albumin in phosphate-buffered saline for 60 min. The cells were immunolabeled with fluorescein isothiocyanate-conjugated CD90 antibodies, and the fluorescence was measured accordingly. The nuclei were counterstained with propidium iodide.
Pluripotency marker analysis of HGMSCs
Pluripotent marker analysis for HGMSCs was performed similarly to multipotent marker analysis, but with a StemLight Pluripotency Antibody Kit (Oct-4, NANOG, SOX-2, SSEA-4, TRA-1-60 TRA-1-81), according to the manufacturer’s instructions.
Encapsulation of HGMSCs in the bioconjugated protein hydrogel (3D)
Encapsulation studies were conducted in two steps. In the first step, bioconjugated protein was prepared according to the patented (Indian-Del No. 1413del2013) procedures, as summarized.11 In brief, gelatin protein was bioconjugated with phenolic acids, and the resultant conjugated protein obtained in the form of powder was used for encapsulation. In the second step, 2 × 106 HGMSCs were mixed with 6–10% conjugated protein in solution and then subjected to gelation using 25–50 μl of sodium periodate. The gelation rate of the protein solution with and without HGMSCs was calculated. The HGMSCs-encapsulated hydrogel was subjected to proliferation and differentiation studies, as described in the following paragraphs.
Cell survival assay of encapsulated HGMSCs
Followed by encapsulation, cell survival was determined using Calcein AM and ethidium bromide live staining11 for different time periods of incubation, 24, 48 and 72 h, and images were captured under a Nikon epi-fluorescence microscope and a Zeiss LSM 510 confocal microscope (Supplementary Video).
3H-Thymidine proliferation assay of encapsulated HGMSCs
3H-Thymidine incorporation and proliferation assay for encapsulated HGMSCs were carried out similarly to the protocol described for free HGMSCs, except for the additional step of treatment with trichloroacetic acid.
Multipotent and pluripotent marker analysis for encapsulated HGMSCs
Encapsulated HGMSCs were maintained in a growth medium αMEM and 10% MSC fetal bovine serum for a period of 7 days. After the scheduled time period, frozen cryosections12 were prepared and subjected to immunofluorescence staining as described above using CD cell-surface markers CD90 and a StemLight Pluripotency Antibody kit (Oct-4, SSEA-4, TRA-1-81). The nuclei were counterstained with 4′,6-diamidino-2-phenylindole, dihydrochloride or propidium iodide, and the images were observed using a Nikon TE2000 Eclipse inverted fluorescence microscope.
Neuronal differentiation studies on HGMSCs in free (2D) and encapsulated forms (3D)
Third-passage cells were used for neuronal differentiation. A two-step induction method consisting of a series of growth factors to induce differentiation into a neuronal lineage13 was followed. In brief, both 2D and 3D HGMSCs were induced for 4 days with step-1 media that contained Dulbecco’s Modified Eagle Medium - F12 (DMEM-F12) supplemented with 5 ng ml−1 of FGF-2, 5 ng ml−1 of nerve growth factor, 2 ng ml−1 of epidermal growth factor, 10 μM of hydrocortisone and 0.1 mM of 3-isobutl-1-methylxanthine. They were then induced for 3 days with step-2 medium containing DMEM-F12 supplemented with 0.5 μM retinoic acid in 3% fetal bovine serum.
Neuronal marker analysis using immunofluorescence
After two steps of neuronal inductions, 2D (free) cells were subjected to preprocessing (as described earlier under multipotent markers analysis by immunoflourescence) for immune labeling with β III tubulin and GFAP, Map-2 and S100 and visualized using a fluorescence microscope. The nuclei were counterstained with DAPI or propidium iodide. With respect to 3D (encapsulated) cells, frozen cryosection was used for immune labeling with the said markers.
Histopathological analysis of encapsulated HGMSCs
Followed by culturing, 3D HGMSCs constructs were fixed for 2 h in formalin solution. The constructs were then transferred and dehydrated with alcohol gradient, paraffin embedded and then sectioned into 5-μm slices using a rotary microtome. The sections were stained with hematoxylin and eosin and cresyl violet staining,14 and the images were captured.
Gene-expression analysis with special reference to transient cells
The gene-expression study was carried out using pluripotency markers Oct-4, Nanog and Sox-2 and neuronal markers β III tubulin, MAP-2 and Nestin13 for 2D HGMSCs and its derived neuronal cells and 3D HGMSCs and its derived neuronal cells using semiquantitative reverse transcriptase–PCR. RNA was isolated using TRIzol reagent (Merck, Genie, Bengaluru, India), as per the manufacturer’s instructions. The RNA pellet was dissolved in 20 μl of molecular biological-grade water, placed at 55 °C for 5 min and measured spectrophotometrically at 260 nm. cDNA was synthesized using a Verso cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, USA). The conversion and cycling program was performed as per the manufacturer’s instructions. cDNA was diluted in a 1:10 ratio, and 3 μl was used for the PCR. The PCR reaction contained 10 pmol of forward and reverse primer (Bioserve, Hyderabad, India) and 2 units of Taq DNA polymerase. To confirm the homogeneity of RNA loading, β-actin was used as a housekeeping gene.
Statistical analysis
All experimental data are represented as the mean±s.d. of the duplicate (n=2) from two different experiments. The statistical significance of the groups was calculated with an independent Student’s t-test using GraphPad Prism; *P<0.05.
Results
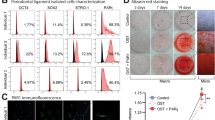
The HGMSCs obtained from the patients were cultured accordingly. Figure 1 depicts the morphological representation of HGMSCs, and the images illustrate the initial culturing studies with respect to the proliferation of HGMSCs, shown in Figures 1a–f. Figure 1g illustrates the percentage level of 3H-thymidine in the HGMSCs of all five patients tested. It was observed that irrespective of the patients, the cells showed proliferation, and no significant difference was observed.

Morphology of cultured gingival cells from gingival tissue. The phase-contrast micrographs of gingival cells on day 1 (a), day 5 (b), day 7 (c) and day 18 (d) of primary culture show a small and large colony composed of densely distributed spindle (black arrow)-shaped cells. On day 15, the cells became flattened and surrounded by proliferating cells and reached confluency. Scale bar =50 μm. Original magnifications × 200. The morphology of the colony formation of HGMSCs represents that P2 cells stained with 1% crystal violet in methanol at day 15 of culture (e, f) show CFU-F colonies. Scale bar=20 cm. Original magnifications × 100. The proliferative effect of HGMSCs in vitro was measured by 3H-thymidine incorporation assay. (g) The amount of incorporated 3H-thymdine was measured after 24 h of culturing. Bars represent the mean±s.e.m. (n=3). The flow cytometry analysis of HGMSCs, passage 3. (h) The expression is strongly positive for MSC-specific surface markers CD105, CD90, CD73 and negative for CD34 and CD45, compared with Isotype control, thus confirming their identity as MSCs. A histogram represents the expression level percentage. The mesenchymal-lineage differentiations of HGMSCs into osteocytes, adipocytes and chondrocytes. In vitro differentiated cells showed positive staining in the specific staining methods. (i) The deposition of a mineralized extracellular matrix (red) was seen after alizarin red staining. Adipogenic differentiation was confirmed by the Oil Red O staining of intracytoplasmic lipid droplets. The chondrogenic differentiation of MSCs with the positive staining of proteoglycans by Alcian blue; scale bar =20 μm. Original magnification × 200.
Figure 1h depicts the results on the phenotypic marker analysis of HGMSCs carried out under flow cytometry analysis. It was observed that all three markers, viz., CD90, CD105 and CD73, were expressed at appreciable levels in comparison with the negative cocktail and the percentages of positive and negative expression, and the histogram represents the expression level of markers. The differentiation studies carried out for adipocytes, osteocytes and chondrocytes are displayed in Figure 1i, suggesting the differentiation of HGMSCs to respective cells in the presence of suitable inducers.
The multi- and pluripotent marker analyses conducted for the obtained HGMSCs are shown in Figure 2a. The cell-surface marker CD90 expressions were observed in HGMSCs at a significant level. In the pluripotent analysis in Figures 2b–g, the expression of Oct-4, Nanog, Sox2, SSEA4 and TRA 160 and 181 observed at substantial levels confirms the pluripotent nature of HGMSCs.

Immunofluorescence staining: The HGMSCs phenotype at passage 3 was determined by immunofluorescence staining. (a) Cells were cultured on glass coverslips and stained with a mouse monoclonal anti-human CD90 IgG (direct fluorescein isothiocyanate conjugated—green), and all nuclei were counterstained with propidium iodide (red). Original magnification × 200 (scale bar =20 μm). HGMSCs were fixed and stained with antibodies against human pluripotency-associated genes (b) Oct-4 (green); (c) Nanog (green); (d) SOX-2 (green); (e) SSEA-4 (green); (f) Tra-1-60 (red) and (g) Tra-1-81 (red) nuclei were counterstained with DAPI. The cells were examined using a Nikon TE Eclipse 2000 immunofluorescence microscope. Positive signals in at least five random high-power fields were observed. The results are representative of at least three independent experiments. Original magnification × 200 (scale=20 μm).
Preparation of 3D HGMSCs-encapsulated hydrogel construct
Figure 3 shows a schematic representation of the CBG hydrogel preparation and encapsulation with HGMSCs and neuronal induction. Figure 4a illustrates the chemistry behind the preparation of bioconjugated protein hydrogel and the phase transition of solution to hydrogel. It was found that the gelation time varied with the concentration of conjugated protein samples used, as shown in Figure 4b. Furthermore, when HGMSCs were incorporated into the conjugated protein solution, followed by gelation, no significant change in the gelation time occurred.

Schematic representation of CBG hydrogel preparation and HGMSCs encapsulation and neuronal differentiation.

(a) The chemistry behind the preparation of bioconjugated protein hydrogel. (b) The histogram shows the gelation time by the tube inversion assay of CBG and CBG+HGMSCs. The values are represented as the mean±s.e.m. of three independent experiments. (c) The 3H-thymidine incorporation assay of encapsulated HGMSCs (3D). The amount of incorporated 3H-thymdine was measured using a Geiger-Muller counter. The histogram represents the mean±s.e.m. n=3. (d, e and f) Representative fluorescence images of CBG hydrogel-encapsulated HGMSCs stained with Calcein AM (live) and ethidium bromide (dead) staining. Live cells were stained green, and dead cells were stained red. Scale bar=100 μm. Original magnification × 100. (g) Confocal Z-stack (300 μm) images of CBG hydrogel-encapsulated HGMSCs stained with Calcein AM (live) and ethidium bromide (dead). Images were collected at 0.27 μm intervals with a 488-nm laser to create a stack in the z-axis. The confocal microscope Z sections were collected at different intervals using sequential excitation for each fluorophore (video and a topographical image of HGMSCs in CBG hydrogel authenticate the presence and survival in the encapsulated form given in the supplementary data). The expression of stem cell markers and pluripotency markers in CBG hydrogel were assessed using immunofluorescence staining. (h) The expression of stem cell markers CD90 (green) counterstained with DAPI (blue). The expression of pluripotency markers by immunofluorescent staining SSEA-4 (green), Tra-1-81 (red) and merged; nuclei counterstained with DAPI (blue). Magnification × 200; scale bar=20 μm.
Followed by encapsulation, HGMSCs were subjected to cell proliferation, cell survival, and multipotent and pluripotent analyses. In addition, 3H-thymidine incorporation analysis (Figure 4c) further confirmed the proliferation of encapsulated HGMSCs. The results demonstrated fluorescence emission upon Calcein and EtBr staining. Observations for 24, 48 and 72 h confirm the well survival of the cells inside the matrix in Figures 4d–f. The z-axis sectioning of the matrix (Figure 4g) and by the use of ImageJ 3D software, the topographical image and video further authenticate the survival of the encapsulated cells, given in the Supplementary File, illustrates the sectioning and the presence of cells. Multipotent and pluripotent analyses of the encapsulated HGMSCs assessed using CD90, SSE4 and TRA 160 demonstrated significant expressions, as shown in Figure 4h. All these results clearly indicated that the functionality of encapsulated HGMSCs was maintained in a similar manner as free cells.
Neuronal differentiation of free and encapsulated HGMSCs
Figures 5a–c illustrate the differentiation of 2D HGMSCs into neuronal lineages, as evidenced with the expression analyses on CD44, β III tubulin, GFAP, S100 and MAP2. Figures 5d and e depict the similar expressions evidenced with the 3D HGMSCs for the aforementioned selected markers.

Immunofluorescence staining of HGMSCs-derived neuronal lineages with the panel of neuronal markers. The cells were stained positively with β III tubulin (green), CD44 (red) and merged images (a). (b) MAP2 (green), β III tubulin. (c) S100 (blue) and GFAP (green) at day 7 show the extent of the differentiation of neurons. Nuclear counterstaining (blue) with DAPI and PI; scale bar=20 μm; original magnification × 200 and × 400. Immunofluorescence staining of HGMSCs-derived neurons in CBG hydrogel (d) S100 (blue), GFAP (green) merged. (e) β III tubulin (green), MAP2 (red) and merged (last column). All nuclei were counterstained with PI (red) and DAPI (blue). Original magnification × 200; scale bar =50 μm.
Furthermore, a histology analysis (H&E staining) of 3D HGMSCs demonstrated the presence of cells with normal characteristic features in the encapsulated form shown in Figures 6A and B. In addition, cresyl staining confirmed the presence of Nissl granule accumulation, which is the characteristic feature of matured and functional neuronal lineages, as illustrated in Figure 6C. Rat hippocampus was used as a positive control, as shown in Figure 6D.

(A) Hematoxylin–eosin staining showing the HGMSCs cultured in CBG hydrogel. (B) Hematoxylin and eosin staining of neuronal inducted cells in in vitro culture (3D). (C) The expression of Nissl bodies of differentiated neuronal cells in CBG hydrogel for staining myelinated axons with (D) a positive control rat hippocampus. Original magnification (A, B) × 400; (C, D) × 200 scale bar=20 μm. (E) The expression of pluripotency markers (Oct-3/4, Nanog and Sox-2) and neuronal markers β III tubulin, nestin and MAP-2 in GMSC third passages were analyzed by RT–PCR. (a) Expression of pluripotency genes (2D). (b) Expression of transcription factors in CBG hydrogel (3D). (c, d) Differentiated neuronal lineages in both 2D and 3D. (e) β-Actin as an internal control. The histogram represents the normalization of gene expression using β-actin as a reference gene. (f) The percentage of gene expression for 2D and (g) the percentage of gene expression for 3D. Values are expressed as the mean±s.d. of three independent experiments. All genes were electrophoretically analyzed on a 1.5% agarose gel and visualized by a gel documentation unit (Bio-Rad, Hercules, CA, USA). A 100-bp DNA ladder was used as a marker.
Figure 6E (a–e) depicts the gene-expression analysis conducted for 2D HGMSCs, 3D HGMSCs and the respective transient levels. It was observed that the 2D HGMSCs expressed all four markers, that is, Oct-4, Nanog, Sox2 and β III tubulin, at an appreciable level with less expression of nestin and nil expression of MAP2. Upon encapsulation, there was no significant difference in the expression patterns of Oct-4, Nanog, Sox2 and β III tubulin, confirming that encapsulation did not affect the basic functional gene expression of HGMSCs. With respect to nestin and MAP2, similarly to free cells, the expressions were feeble and nil, respectively, in the 3D cells. In the case of transient cells, we found that the expression of Oct-4 was reduced completely, the Nanog expression was meager, and SOX2 and β III tubulin expressions were maintained. Notably, nestin and MAP2 expressions were found to be high in transient cells, indicating the maintenance of the functional properties of neurons at a significant level in both free and encapsulated forms. Percentage levels of gene expression are shown in Figure 6E (f and g).
Discussion
The current scenario on stem cell therapy necessitates much intensive research on (i) an alternative to birth-associated tissues and (ii) carrier material to support differentiation at the expected levels when translational studies are executed. The demand for neuronal regeneration further intensifies research initiatives. With respect to the alternatives, oral stem cells, which are of neural crest origin, are found to be suitable due to their potency and availability. Compared with other oral stem cells, gingiva may be an attractive candidate and be considered for research on stem cell therapy. In addition to their mechanical strength and biocompatibility, the ‘bio-mimetic’ nature (physico-chemical similarity to the native extracellular matrix) of hydrogel gives them an edge over other scaffolds in biological applications.15 Natural polymers such as collagen, hyaluronate and gelatin, despite being biomimetic, are weak candidates as scaffolds for regeneration due to two major factors: a lack of mechanical strength and a lack of homogeneity in drug or cells when loaded. Even with hydrogel, compared with conventional fabrication of hydrogel, techniques such as injectable hydrogel are more effective in gaining mechanical strength and homogeneity/uniform drug or cell dispersal when added.16
The hydrogel used in the present study is a combination of caffeic acid and gelatin. In brief, dihydroxy phenolic acid (caffeic acid) was conjugated with gelatin, and the oxidation of phenolics using mild oxidizing agents transformed the conjugated protein into a hydrogel.11 In vitro and in vivo assessments of a 3-(3, 4-dihydroxyphenyl)-2-propenoic acid-bioconjugated gelatin-based injectable hydrogel were conducted for biomedical applications. Caffeic acid (3-(3,4-dihydroxyphenyl)-2-propenoic acid) is a plant phenolic widely present in, for example, vegetables, fruits, coffee, tea, olive oil and red wine.17
This yellow solid consists of hydroxyl and a, b-unsaturated (acrylic) carboxylic groups. In wound healing, caffeic acid exhibits antimicrobial, antioxidant and anti-inflammatory actions and attenuates enzymes such as matrix metalloproteinase (MMP and 9), lipoxygenase and cyclooxygenase.18 In fibroblast cell lines, caffeic acid has the least cytotoxicity among the various phenolic compounds.17 Gelatin can be crosslinked or modified with the inclusion of other materials to significantly alter its mechanical and biochemical properties, and it is used in the preparation of bioadhesives, scaffolds, cell-sheet carriers and hydrogels. Raja et al.11 demonstrated that gelatin has a greater storage modulus than the loss module, suggesting the elastic nature of this hydrogel. Additionally, a weight gain of approximately 50% within 12 h during swelling studies and a weight loss of approximately 50% within 12 h during evaporation suggested the suitability of the CBG gel as a drug carrier. The drug carried in this hydrogel had an initial burst and a later sustained release, which we felt could be beneficial when applied for regeneration, where the initial release of cells could augment regeneration. This highly biocompatible hydrogel promotes cell migration and exhibits radical scavenging behavior.
The preparation procedure followed in the present study involved in situ gelation, which is considered to be the most important criterion for the encapsulation of cells inside the matrix. The loss of cells during encapsulation was highly minimized. The presence of cells was confirmed through immunostaining, and the observations of proliferation (3H-thymidine assay) provided encouraging results to proceed further.
With respect to differentiation, techniques such as the addition of growth factors including bone morphogenetic protein, basic fibroblast growth factor, retinoic acid,13 micro-RNA transfection19 and scaffold elasticity were followed and reported in the literature. However, in the present study, we employed conventional basic fibroblast growth factor and retinoic acid for induction. Comparing the differentiation factors alone or in combination, we found that basic fibroblast growth factor and retinoic acid combination expressed the highest level of neuronal markers, which could be the reason for the significant expression of many of the markers representing self-renewing stem cells to well-differentiated neurons or astrocytes in our study (Table 1).
According to Foudah et al., even undifferentiated mesenchymal cells from different sources, such as bone marrow, periodontal sources and pulpal sources, spontaneously express neural markers such as β III tubulin and Neu N, which prompted us to use more markers to confirm the neuronal cells.20 Notably, the authors also observed that the expression of nestin was greater with periodontal and pulpal stem cells than the cells of bone and adipose origins due to the neural crest origin of the periodontal and pulpal cells. The high expression of nestin observed in the present study strongly supports the neural crest origin of gingiva.2
With reference to neural stem cell differentiation, these cells are divided into progenitors, glial-restricted progenitors and neuronal-restricted progenitors. Under the influence of factors, the former can turn into either astrocytes or oligodendrocytes. Astrocytes are star shaped and provide biochemical support to endothelial cells that form the blood–brain barrier. Oligodendrocytes lend support to neurons, and neuronal-restricted progenitors differentiate into neurons.21 Thus, with available markers, the pluripotency status, the progenitor condition of neuronal and glial stem cells, and finally the differentiated neurons and astrocytes were confirmed.
In using encapsulated cells for cell regeneration and differentiation, as summarized, the extracellular matrix surrounding the cell is important for growth, development and cell functioning. In the present study, the use of bioconjugated hydrogel, that is, caffeic acid-bioconjugated gelatin hydrogel, for HGMSCs demonstrated appreciable survival, proliferation and differentiation. Because the hydrogel has a substantial storage modulus (1800 Pa) in addition to hydrophilicity and an appreciable water-holding capacity, it helps HGMSCs survive and proliferate.
Notably, the observations of differentiation into the neuronal lineages of free cells and encapsulated cells showed a significant level of gene expression markers in the encapsulated cells, suggesting that the native functional and mechanical properties of the chosen hydrogel may stimulate the differentiation process. In addition, we found that the conjugated hydrogel contains 10–15% of free phenolic upon crosslinking, and the inducing effect of phenolic acids may be exerted in the differentiation. However, more explorations are needed to substantiate the inducing effect of phenolic acids in the differentiation of mesenchymal cells. The existing interconnected pores facilitate the mobility of cells and nutrients substantially. No toxicity issues for the encapsulated HGMSCs were observed, and the appreciable biocompatibility suggested the suitability of the bioconjugated hydrogel for cell22 differentiation, followed by therapeutic applications.
A revolutionary mechano-biology concept introduced by Engler et al.23 suggested the importance of the modulus of elasticity in stem cell regeneration and the modulus with 1 kPa favored neuronal lineages.22 Thus, the observed storage modulus of the bioconjugated hydrogel may also be responsible for the differentiation of HGMSCs to neuronal cells. The histological section of the encapsulated cell when stained with cresyl violet demonstrated the presence of Nissl bodies, and the immunohistochemistry of the cryosection showed positivity for CD90, SSEA4, 1-81, β III tubulin, MAP2, GFAP and S100, confirming the status of stem and neuronal cells. The vitality of the stem cells was maintained in the hydrogel, as evidenced by the confocal z-axis sectioning. The thymidine incorporation assay suggested an appreciable level of thymidine, indicating the marginal increase in the proliferation of the differentiated cells.
There was no significant increase or decrease in the expression of selected markers such as Oct-4, Nanog, Sox2, Nestin, β III tubulin or MAP2, suggesting that the encapsulated cells in the gel were in different stages. Oct-4 and Sox2 are transcription factors that maintain embryonic stem cell pluripotency24 and are expressed in an undifferentiated state of MSCs. At a transient level, Oct-4 and Nanog expression regressed towards neuronal lineages. Sox2 was upregulated, and its expression levels were maintained throughout neuronal differentiation, suggesting that Sox2 plays a key role in neural differentiation.25 Similarly, our results show a significant decrease in the expression levels of Oct-4 and Nanog and an increased level of Sox2 in differentiated neuronal cells. Neuronal progenitor Nestin, β III tubulin and MAP2 (maturation neuronal markers) showed a significantly elevated expression in a differentiated state, suggesting that encapsulated HGMSCs mimic the tissue-like construct and provide an environment for maintaining neuronally derived cells and their functional properties.
In conclusion, this study exemplifies the differentiation of HGMSCs to neuronal lineages under 2D (free) and 3D (encapsulated) forms. Differentiation was confirmed through immunostaining with selective gene expression markers. The functional and mechanical stability of hydrogel supports cell survival and proliferation, as evidenced by thymidine incorporation and immunostaining. A new tissue engineering 3D construct of HGMSCs encapsulated in hydrogel with all the requisite functional properties may be a viable functional construct in stem cell therapy.
References
Marynka-Kalmani K, Treves S, Yafee M, Rachima H, Gafni Y, Cohen MA et al. The lamina propria of adult human oral mucosa harbors a novel stem cell population. Stem Cells 2010; 28: 984–995.
Xu X, Chen C, Akiyama K, Chai Y, Le AD, Wang Z et al. Gingivae contain neural-crest- and mesoderm-derived mesenchymal stem cells. J Dent Res 2013; 92: 825–832.
Engler AJ, Sen S, Sweeney HL, Discher DE . Matrix elasticity directs stem cell lineage specification. Cell 2006; 126: 677–689.
Cai L, Dewi RE, Heilshorn SC . Injectable hydrogels with in situ double network formation enhance retention of transplanted stem cells. Adv Funct Mater 2015; 25: 1344–1351.
Williams AF, Gagnon J . Neuronal cell Thy-1 glycoprotein: homology with immunoglobulin. Science 1982; 216: 696–703.
Gao Y, Zhao G, Li D, Chen X, Pang J, Ke J . Isolation and multiple differentiation potential assessment of human gingival mesenchymal stem cells. Int J Mol Sci 2014; 15: 20982–20996.
Hartley RS, Yablonka-Reuveni Z . Long-term maintenance of primary myogenic cultures on a reconstituted basement membrane. Vitr Cell Dev Biol 1990; 26: 955–961.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006; 8: 315–317.
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999; 284: 143–147.
Solchaga LA, Penick KJ, Welter JF . Chondrogenic differentiation of bone marrow-derived mesenchymal stem cells: tips and tricks. Methods Mol Biol 2011; 698: 253–278.
Thirupathi Kumara Raja S, Thiruselvi T, Aravindhan R, Mandal AB, Gnanamani A . In vitro and in vivo assessments of a 3-(3,4-dihydroxyphenyl)-2-propenoic acid bioconjugated gelatin-based injectable hydrogel for biomedical applications. J Mater Chem B 2015; 3: 1230–1244.
Janes KA, Wang C-C, Holmberg KJ, Cabral K, Brugge JS . Identifying single-cell molecular programs by stochastic profiling. Nat Methods 2010; 7: 311–317.
Divya MS, Roshin GE, Divya TS, Rasheed VA, Santhoshkumar TR, Elizabeth KE et al. Umbilical cord blood-derived mesenchymal stem cells consist of a unique population of progenitors co-expressing mesenchymal stem cell and neuronal markers capable of instantaneous neuronal differentiation. Stem Cell Res Ther 2012; 3: 57.
Shafri MAM, Jais AMM, Jaffri JM, Kim MK, Ithnin H, Mohamed F . Cresyl violet staining to assess neuroprotective and neuroregenerative effects of haruan traditional extract against neurodegenerative damage of ketamine. Int J Pharm Pharm Sci 2012; 4: 163–168.
Bae KH, Wang L-S, Kurisawa M . Injectable biodegradable hydrogels: progress and challenges. J Mater Chem B 2013; 1: 5371.
Li Y, Rodrigues J, Tomás H . Injectable and biodegradable hydrogels: gelation, biodegradation and biomedical applications. Chem Soc Rev 2012; 41: 2193–2221.
Koganov MM, Dueva OV, Tsorin BL . Activities of plant-derived phenols in a fibroblast cell culture model. J Nat Prod 1999; 62: 481–483.
Wu W, Lu L, Long Y, Wang T, Liu L, Chen Q et al. Free radical scavenging and antioxidative activities of caffeic acid phenethyl ester (CAPE) and its related compounds in solution and membranes: a structure–activity insight. Food Chem 2007; 105: 107–115.
Zhou Y, Chen K-S, Gao J-B, Han R, Lu J-J, Peng T et al. miR-124-1 promotes neural differentiation of rat bone marrow mesenchymal stem cells. Zhongguo Dang Dai Er Ke Za Zhi 2012; 14: 215–220.
Foudah D, Monfrini M, Donzelli E, Niada S, Brini AT, Orciani M et al. Expression of neural markers by undifferentiated mesenchymal-like stem cells from different sources. J Immunol Res 2014; 2014: 987678.
Holland EC . Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet 2001; 2: 120–129.
Leipzig ND, Shoichet MS . The effect of substrate stiffness on adult neural stem cell behavior. Biomaterials 2009; 30: 6867–6878.
Tse JR, Engler AJ . Preparation of hydrogel substrates with tunable mechanical properties. Curr Protoc Cell Biol 2010; 10: 1–16.
Lu Y, West FD, Jordan BJ, Mumaw JL, Jordan ET, Gallegos-Cardenas A et al. Avian-induced pluripotent stem cells derived using human reprogramming factors. Stem Cells Dev 2012; 21: 394–403.
Noisa P, Ramasamy TS, Lamont FR, Yu JSL, Sheldon MJ, Russell A et al. Identification and characterisation of the early differentiating cells in neural differentiation of human embryonic stem cells. PLoS ONE 2012; 7: e37129.
Acknowledgements
We thank the Ministry of Micro, Small and Medium Enterprises (MSME) for the use of the Geiger-Muller counters for the proliferation study.
Author contributions
RRS conceived of the concept, performed clinical procedures, analysis, interpretation and a critical review, and drafted the manuscript. RS contributed to the design, performed the experiments and data acquisition, and drafted the manuscript. Both SR and RRS contributed equally to this work. MGD contributed to the performance of the experiments, critically revised the manuscript and interpreted the data. AG contributed to the hydrogel scaffold preparation, participated in discussion and revised the manuscript. STKR contributed to the hydrogel preparation and reviewed the manuscript. All authors gave their final approval and agree to be accountable for all aspects of the work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Rao, S., Subbarayan, R., Dinesh, M. et al. Differentiation of human gingival mesenchymal stem cells into neuronal lineages in 3D bioconjugated injectable protein hydrogel construct for the management of neuronal disorder. Exp Mol Med 48, e209 (2016). https://doi.org/10.1038/emm.2015.113
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2015.113
This article is cited by
-
Insulin-producing cell clusters derived from human gingival mesenchymal stem cells as a model for diabetes research
Molecular Biology Reports (2022)
