
Abstract
New colchicine analogs have been synthesized with the aim of developing stronger potential anticancer activities. Among the analogs, CT20126 has been previously reported to show immunosuppressive activities. Here, we report that CT20126 also shows potential anticancer effects via an unusual mechanism: the modulation of microtubule integrity and cell cycle arrest at the G2/M phase before apoptosis. When we treated COS-7 cells with CT20126 (5 μM), the normal thread-like microtubules were disrupted into tubulin dimers within 10 min and thereafter repolymerized into short, thick filaments. In contrast, cells treated with the same concentration of colchicine exhibited microtubule depolymerization after 20 min and never underwent repolymerization. Furthermore, optical density (OD) analysis (350 nm) with purified tubulin showed that CT20126 had a higher repolymerizing activity than that of Taxol, a potent microtubule-polymerizing agent. These results suggest that the effects of CT20126 on microtubule integrity differ from those of colchicine: the analog first destabilizes microtubules and then stabilizes the disrupted tubulins into short, thick polymers. Furthermore, CT20126 induced a greater level of apoptotic activity in Jurkat T cells than colchicine (assessed by G2/M arrest, caspase-3 activation and cell sorting). At 20 nM, CT20126 induced 47% apoptosis among Jurkat T cells, whereas colchicine induced only 33% apoptosis. Our results suggest that the colchicine analog CT20126 can potently induce apoptosis by disrupting microtubule integrity in a manner that differs from that of colchicine or Taxol.
Similar content being viewed by others

Introduction
Microtubules, which are structural components of the cytoskeleton, are composed of α- and β-tubulin heterodimers and play key roles in many biological events, including the development and maintenance of cell shape, intracellular transport, cell division and cell growth.1, 2 A subset of microtubules called spindle fibers support proper cell division by pulling the chromosomes to either side of the dividing cell. In the search for cell growth inhibitors with the potential for use as anticancer therapies, many inhibitors of microtubule dynamics have been investigated, including colchicine and paclitaxel (Taxol).3, 4, 5, 6, 7 Taxol binds and stabilizes microtubules, thereby suppressing their dynamic rearrangements, which are necessary for cell growth.8 In contrast, colchicine is a Vinca alkaloid that binds to tubulin and inhibits its polymerization by blocking the cell cycle at the G2/M phase and triggering apoptosis.9, 10 Such an inducer of apoptosis was seen as a strong candidate for anticancer therapy, and numerous papers have reported colchicine-induced apoptosis in many cell types. However, colchicine was found to have some toxic effects, including neuropathy.11, 12 Thus, numerous researchers have synthesized and tested a variety of colchicine derivatives in the hope of developing an agent with improved efficacy and decreased side effects.
Colchicine is also widely used as an immunosuppressant during kidney transplantation.13, 14 In recent years, our group has synthesized many colchicine derivatives and assessed their immunosuppressant potential. Among them, the semisynthetic colchicine derivative CT20126 was found to act as an effective immunosuppressant.15, 16 While examining the effects of CT20126, we noted that it triggered drastically different microtubule dynamics and apoptotic activity compared with colchicine. Cells treated with CT20126 resulted in an increase of sub-G1 population and caspase-3 activation, which suggests that CT20126 has potent apoptotic activity. In the current study, we examined the effects of CT20126 on microtubule activity and apoptosis and compared them with the effects of colchicine in terms of cancer therapeutics.
Materials and methods
Reagents
Monoclonal anti-β-tubulin and anti-α-tubulin were purchased from Chemicon (Temecula, CA, USA) and Sigma-Aldrich (St Louis, MO, USA), respectively. Polyclonal anti-β-tubulin and monoclonal anti-acetylated α-tubulin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit antibodies were purchased from Upstate (Lake Placid, NY, USA). Highly purified tubulin from bovine brain was obtained from Sigma-Aldrich. The fluorescein-conjugated Affinipure goat anti-rabbit immunoglobulin G (IgG) and rhodamine-conjugated Affinipure goat anti-mouse IgG were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Colchicine and paclitaxel (Taxol) were obtained from Sigma-Aldrich, and CT20126 was kindly provided by Chem Tech Research Incorporated (CTRI, Whasung, Korea). Colchicine, Taxol and CT20126 were dissolved in dimethyl sulfoxide and stored at −20°C.
Cell culture
COS-7 cells were cultured at 37 °C in Dulbecco’s modified Eagle’s medium that contained 10% fetal bovine serum, 5% horse serum and 100 U of penicillin–streptomycin. Jurkat T cells were cultured in RPMI-1640 medium that was supplemented with 10% fetal bovine serum and 100 U of penicillin–streptomycin.
In vitro assay of tubulin assembly
Purified tubulin (0.1 μg μl−1) was centrifuged at 15 000 × g for 30 min, and the supernatants were equilibrated in tubulin polymerizing buffer (0.1 M MES, 1 mM EGTA, 0.5 mM MgCl2, 0.1 mM EDTA and 2.5 M glycerol, pH 6.5) on ice.17 The reaction was initiated by the addition of 1 mM guanosine-5'-triphosphate (GTP) at 37 °C in a 200-μl reaction volume in a light path cell and monitored using a thermostatic spectrophotometer at 350 nm. After 20 min, the samples were mixed with 5 nM CT20126, colchicine or Taxol and incubated at 37 °C for 5 min. The samples were cooled in an ice bath for depolymerization (still within the light path cell). Successive reactions were performed for another 25 min at 37 °C.
Immunofluorescent microscopy
COS-7 cells were seeded on glass coverslips in 6-well plates and grown for 2 days in Dulbecco’s modified Eagle’s medium that was supplemented with 10% fetal bovine serum. For CT20126 treatment, the cells were serum starved for 24 h before incubation with 5 μM CT20126. After the appropriate times, the cells were fixed at 37 °C for 10 min in 4% paraformaldehyde and then incubated with affinity-purified anti-α or β-tubulin antibodies for 1 h at room temperature in a humidified chamber. Following a complete washing with phosphate-buffered saline, the cells were incubated with rhodamine-conjugated Affinipure goat anti-mouse IgG. Immunostained cells were observed with a fluorescent microscope (Eclipse E600 Epifluorescence Microscope; Nikon Corporation, Tokyo, Japan), and the images were captured with a digital microscopic camera.
Western blot analysis
The levels of α-tubulin acetylation and PARP (poly (ADP-ribose) polymerase) degradation (reflecting caspase-3 activity) were determined by western blot analysis. Approximately 6 × 106 Jurkat T cells were harvested by centrifugation, lysed and resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The resolved proteins were transferred to a nitrocellulose membrane, and the blot was hybridized with anti-acetylated α-tubulin antibody or anti-PARP antibody. The acetylated α-tubulin and 85-kDa PARP fragments were detected with a horseradish peroxidase-conjugated secondary antibody using an ECL detection system (GE Healthcare, Wauwatosa, WI, USA).
Cell counting and flow cytometric analysis
Cell counts were performed using a hemocytometer. Approximately 1 × 106 Jurkat T cells were suspended in 100 μl of phosphate-buffered saline and vortexed with 200 μl of 95% ethanol. The cells were incubated at 4 °C for 1 h, washed with phosphate-buffered saline, resuspended in 250 μl of 1.12% sodium citrate buffer (pH 8.4) containing 12.5 μg of RNase and incubated at 37 °C for 30 min. The cellular DNA was stained with 250 μl of propidium iodide (50 μg ml−1) for 30 min at room temperature. The stained cells were analyzed by fluorescence-activated cell sorting (FACS) on a FACScan flow cytometer, and relative DNA contents were assessed based on red fluorescence.18
Results
In vitro microtubule assembly by CT20126
The effect of CT20126 on in vitro microtubule assembly in a tubulin assembly buffer (0.1 M MES, 1 mM EGTA, 0.5 mM MgCl2, 0.1 mM EDTA, 1 mM GTP, 2.5 M glycerol and 0.1 μg μl−1 tubulin, pH 6.5) was measured by assessing the turbidimetric time course of microtubule assembly at 350 nm. Because CT20126 is a colchicine analog, it is expected to show basal microtubule-disrupting activity in the turbidimetric analysis (Figure 1a). However, CT20126 showed microtubule-stabilizing activity rather than microtubule disruption. Figure 1b shows that the tubulin-polymerizing activity of CT20126 was significantly higher than that of the highly active microtubule-stabilizing drug Taxol.

Turbidity-assessed time course of in vitro microtubule assembly. (a) Chemical structures of colchicine and CT20126. (b) Taxol, colchicine or CT20126 (5 nM each) were added to tubulin polymerizing buffer containing 0.1 μg μl−1 tubulin and 1 mM guanosine-5'-triphosphate (GTP; arrow ‘a’). After the samples were cooled on an ice bath for depolymerization, repolymerization was initiated by increasing the temperature to 37 °C (arrow ‘b’). DMSO, dimethyl sulfoxide. (c) Dose-response effects of CT20126 on microtubule polymerization. Arrows ‘a’ and ‘b’ indicate the drug treatment and rewarming times, respectively.
We also examined the dose-response effects of CT20126 on tubulin polymerization. Figure 1c shows that nanomolar concentrations of CT20126 increased the OD of the polymerized tubulin, which suggests that CT20126 accelerated tubulin polymerization at this dose. To avoid the possibility that the high OD in the presence of CT20126 was due to the formation of amorphous aggregates of tubulin in vitro, we examined the polymerization process through cooling and warming cycles. As described in the ‘Materials and methods’ section, purified tubulin was polymerized in the presence of 1 mM GTP at 37 °C. After the turbidity plateau had been reached, the polymerized tubulin was depolymerized by incubation on ice for 5 min. Figures 1b and c show that the cooling step resulted in complete depolymerization (OD 350 nm<0.1), even in the presence of CT20126. Rewarming at 37 °C increased the OD, which suggests that the repolymerization reaction was sped up in the presence of CT20126. These results indicate that the synthetic colchicine derivative CT20126 induces stabilization of tubulin.
Microtubule assembly in cultured cells treated with CT20126
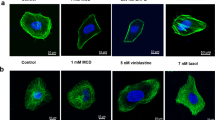
Because CT20126 stabilized microtubules in vitro, we examined its effects on microtubules in living cells. First, we examined the effects of various (in μM) concentrations of CT20126 on the cytoplasmic microtubules of COS-7 cells by assessing the immunocytochemical staining with an anti-β-tubulin antibody (Figure 2a). In contrast to the in vitro tubulin polymerization, microtubule modulation of cultured COS-7 cells required a μM concentration of CT20126. The polymerization/depolymerization phenotypes clearly differed between the cultures treated with 1 μM CT20126 and those treated with 10 μM CT20126. Figure 2a shows that the undulating microtubules of untreated COS-7 cells changed to short, thick filaments in the presence of CT20126. We also examined the time courses of cytoplasmic microtubule changes that were induced by CT20126, colchicine and Taxol. Figure 2b shows that 5 μM colchicine completely disrupted the microtubules within 20 min, whereas Taxol triggered a robust and time-dependent stabilization of microtubules. In contrast, CT20126 initially disrupted the cytoplasmic microtubules (within 10 min) and thereafter induced repolymerization of the tubulin, first into numerous glittering spots under the fluorescent microscope and then into short, thick filamentous polymers. Figure 2b shows that these were not simple aggregates because they polymerized from each spot in the presence of CT20126.

Modulation of cellular microtubules in response to CT20126 treatment. (a) COS-7 cells were cultured in the presence of various concentrations of CT20126 for 40 min at 37 °C and processed for immunocytochemistry. (b) Comparisons of cellular microtubule modulation induced by treatment with CT20126, colchicine or Taxol (5 μM each).
Acetylated α-tubulin is more resistant to microtubule depolymerization under depolymerizing conditions and is abundant in cells treated with Taxol;19, 20 thus, tubulin acetylation is considered to be a marker of microtubule stabilization.4, 19, 20 We examined the levels of acetylated tubulin in cells treated with CT20126 or Taxol. COS-7 cells treated with CT20126, Taxol or colchicine were lysed, and the amounts of acetylated tubulin were analyzed using western blotting with an anti-acetylated α-tubulin antibody. Figure 3a shows that the microtubules of CT20126- and Taxol-treated cells were highly acetylated, whereas colchicine-treated cells were less acetylated than the CT20126-treated cells. We observed a threefold increase in response to incubation with 5 μM CT20126 for 1 h (Figure 3b). These results support the notion that microtubules are stabilized by treatment with CT20126 or Taxol.

Tubulin acetylation by CT20126. (a) COS-7 cells treated with CT20126 were lysed and immunoblotted with anti-acetylated α-tubulin antibody. (b) The relative intensities of acetylated α-tubulin obtained from the immunoblot in (a).
G2/M arrest of cultured cells by CT20126
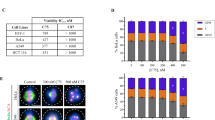
To analyze the effects of CT20126 on apoptosis and the cell cycle, we used flow cytometry to test the cell cycle parameters of Jurkat T cells that were treated with various concentrations of CT20126, colchicine or Taxol (Figure 4a). Jurkat T cells were used because CT20126 has shown immunomodulatory effects and is considered to be a candidate immunosuppressant.15, 16 Figure 4a shows that significant mitotic arrest was observed in response to 5 nM CT20126 and Taxol, whereas colchicine had less of an effect on G2/M arrest. In cultures treated with 10 nM CT20126, 73% of the cells accumulated in the G2/M phase over 24 h, which suggests that apoptosis occurred (Figure 4c). To confirm these findings, we monitored the induction of apoptosis by FACS analysis of the appearance of the sub-G1 peak (Figure 4a). Figure 4b shows that the apoptotic rate gradually increased with the drug concentration. In cultures treated with 20 nM CT20126, almost half of the cells underwent apoptosis (Figure 4b), and Taxol and colchicine triggered ∼30–35% apoptosis. In cultures treated with Taxol, 5 nM showed the highest rate of apoptosis (Figure 4b).

Effects of CT20126 on cell cycle distribution. (a) Representative flow cytometric analyses of Jurkat T cells treated with CT20126, colchicine or Taxol. Jurkat T cells were incubated with various concentrations of CT20126 for 24 h. The cells were then fixed and stained with propidium iodide and analyzed by flow cytometry. (b) The percentage of apoptosis (sub-G1 population) induced by different concentrations of CT20126, colchicine or Taxol. (c) Cell cycle distributions of Jurkat T cells treated with CT20126, colchicine or Taxol. Jurkat T cells treated with different concentrations of each drug for 24 h were analyzed for cell cycle by flow cytometry, and the portions of cells in each phase of the cell cycle were quantified. The data are shown as the means of three independent experiments±s.d.
Consistent with our observation that COS-7 cells that were treated with CT20126 showed increased α-tubulin acetylation (Figure 3), which is considered to be a marker of microtubule stabilization and mitotic arrest,4, 20 5 μM of each drug triggered a clear increase in acetylated tubulin over time, whereas untreated control cells did not show a clear increase (Figure 3).
Involvement of caspase-3 activation in CT20126-induced apoptosis
The acetylation of α-tubulin, which is caused by the inhibition of histone deacetylase 6 (HDAC6),21, 22 leads to the activation of caspase-3 (a member of the cysteine protease family). Notably, caspase activity is required for cellular apoptosis. Western blotting showed that CT20126 induced both caspase-3 activation and the cleavage of the caspase-3 substrate PARP from its 116 kDa form to its 85 kDa fragment (Figure 5a). Moreover, cleavage of the 60 kDa fragment from the 145 kDa intact phospholipase C-γ1 (PLC-γ1), which is another marker for caspase activation,23 was detected by western blotting of CT20126-treated Jurkat T cells with an anti-PLC-γ1 antibody (Figure 5b).

The effects of CT20126 on PARP (poly (ADP-ribose) polymerase) and (PLC-γ1 (phospholipase C-γ1)) cleavage. (a) Jurkat T cells were cultured with different concentrations of CT20126 for 24 h at 37 °C, and cell lysates were analyzed by western blotting with an anti-PARP antibody that detected the 85 kDa fragment of cleaved PARP. (b) Time course of PLC-γ1 cleavage. Jurkat T cells were cultured in the presence of 10 nM CT20126 for 0, 3, 6, 12 and 24 h, and lysates were analyzed by western blotting for the 60 kDa fragment of cleaved PLC-γ1.
Discussion
In the present study, we characterized the effects of CT20126, a colchicine derivative, on microtubule dynamics and apoptosis. Colchicine is known as a microtubule-disrupting agent, but our semisynthetic compound, CT20126, showed an interesting function in its ability to rearrange microtubules. Our data indicate that (1) CT20126 initially disrupts microtubules; (2) unlike colchicine, CT20126 subsequently repolymerizes the disrupted tubulin molecules into thick filaments that do not resemble normal microtubules; (3) CT20126 induces α-tubulin acetylation; and (4) CT20126 triggers apoptosis in association with caspase-3 activation and G2/M phase cell cycle arrest. These data demonstrate a series of events leading from microtubule disruption to apoptosis, which collectively suggest that CT20126 could be a possible candidate for use in anticancer therapy.
CT20126 showed a polymerizing (rather than microtubule-disrupting) effect in a turbidimetric analysis in vitro (Figure 1b). CT20126 acted on purified tubulin (0.1 μg μl−1) at nM concentrations and increased the OD in a dose-dependent manner (Figure 1c). At a higher concentration of tubulin (∼1 μg μl−1), the turbidity of polymerized tubulin within CT20126-treated reactions became too high for successful OD measurement. Several previous in vitro studies have found that colchicine-liganded tubulin forms abnormal polymers in the presence of excess amounts of both tubulin (over 1 μg μl−1) and colchicine (1 mM).24, 25 In contrast, CT20126 appears to act differently because the nM concentrations of CT20126 induced high ODs with a low concentration of tubulin (0.1 μg μl−1; Figure 1c). This observation led us to investigate the dynamics of microtubule instability within cultured cells. CT20126 treatment of COS-7 cells triggered an initial complete disruption of microtubules, which was followed by the repolymerization of tubulin into short, thick filaments (Figure 2a). This suggests that CT20126 produces rapid bundling of repolymerized tubulin, which has previously been shown for bacterial pneumolysin.26 Numerous spots representing the initial repolymerization appeared 30 min after cells were treated with 5 μM CT20126, and extensive bundling or thickening of the filaments was observed 10 min later (Figure 2b). These bundled filaments showed abnormalities (for example, twisting, distortion and shortness) compared with normal microtubules (Figure 2b). Notably, this response required a relatively high concentration (μM level) of CT20126 compared with the concentrations used for our in vitro polymerization experiments. In vitro tubulin polymerization experiments with purified tubulin required nM concentrations of CT20126, whereas μM concentrations were required in living cells. Generally, cellular microtubules are ∼25 nm in diameter and many micrometers in length. In contrast, the abnormal filaments that were formed in the CT20126-treated cells were ∼10- to 20-fold thicker than normal microtubules. This suggests that CT20126 stabilizes tube length (polymerization) during the initial stages and increases tube diameter (bundling) from the completely disrupted microtubule during the later stages. This stabilizing effect was confirmed by the significant increase of acetylated α-tubulin in the COS-7 cells that were treated with CT20126 and assessed via western blotting (Figure 3). Although 5 μM CT20126 required 10 min to disrupt microtubules and another 10–20 min to repolymerize the bundled filaments (30 min total; Figure 2), tubulin acetylation was already detected at 10 min after treatment (Figure 3). This early α-tubulin acetylation may be due to CT20126-induced suppression of microtubule dynamics. There have been several reports of increased α-tubulin acetylation in cells under depolymerizing conditions, and colchicine-resistant microtubules have been found to contain more acetylated α-tubulin than colchicine-sensitive cells.19, 20, 27 In addition, α-tubulin acetylation is induced by the inhibition of HDAC621, 22 and is considered to be a marker of apoptosis.28, 29, 30 Thus, we speculate that CT20126 might directly inhibit HDAC6 activity to result in an early increase in tubulin acetylation. We are currently examining the effect of CT20126 on HDAC6 activity.
In addition to α-tubulin acetylation, there is another possible explanation for microtubule stabilization by CT20126. Colchicine depolymerizes microtubules and blocks mitosis at high concentrations but stabilizes microtubule dynamics at low concentrations.31, 32 At low concentrations, colchicine-bound tubulin heterodimers (colchicine–tubulin complex) copolymerize into a growing microtubule lattice, and the subsequently incorporated colchicine molecules suppress microtubule instability (or stabilize microtubule dynamics).32 In this context, we speculate that the colchicine analog CT20126 has a potent activity for microtubule stabilization via the copolymerization of the tubulin–CT20126 complex into a growing microtubule. This conclusion is consistent with our data from the in vitro and in vivo studies that suggest CT20126 stabilizes microtubule dynamics.
Cell cycle analysis showed that >70% of the cells that were treated with 10 nM CT20126 accumulated at the G2/M phase, whereas both colchicine and Taxol induced a smaller number of cells (Figure 4c). Flow cytometric analysis indicated that 16, 27 and 47% of Jurkat T cells underwent apoptosis within 24 h following treatment with 5, 10 and 20 nM CT20126, respectively (Figure 4b). Moreover, treatment of U937 cells (a human lymphoma cell line) with 10 nM CT20126 induced apoptosis among 83% of cells within 24 h (data not shown). It is well known that the accumulation of the sub-G1 population represents the cell death phase, which includes apoptosis and necrosis. However, caspase activation and genomic DNA fragmentation are specific signals for apoptosis but not for necrosis.33, 34 PLC-γ1 and PARP are well-known substrates for caspase-3, and they produce 60 and 85 kDa fragments by caspase-3 activity.23, 33, 34 CT20126 clearly showed the above apoptotic signals, including PARP and PLC-γ1 degradation (Figures 5a and b). The ability of a candidate agent to induce selective apoptosis in cancer cells is critical to the development of anticancer drugs. Based on the ability of CT20126 to induce apoptosis, CT20126 should be considered a candidate anticancer drug.
In summary, the present study demonstrates that our previously developed colchicine derivative, CT20126, has strong potential as a microtubule modulator and inducer of apoptosis in cancer cells and should be considered as a potential anticancer agent. Future studies into the mechanism by which CT20126 disrupts and repolymerizes microtubules might facilitate the design of better anticancer drugs.
References
Jordan MA, Wilson L . Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004; 4: 253–265.
Hadfield JA, Ducki S, Hirst N, McGown AT . Tubulin and microtubules as targets for anticancer drugs. Prog Cell Cycle Res 2003; 5: 309–325.
Tian W, Qin L, Song Q, He L, Ai M, Jin Y et al. A novel synthetic analog of 5, 8-disubstituted quinazolines blocks mitosis and induces apoptosis of tumor cells by inhibiting microtubule polymerization. PLoS One 2010; 5: e10499.
Mohan R, Panda D . Kinetic stabilization of microtubule dynamics by estramustine is associated with tubulin acetylation, spindle abnormalities, and mitotic arrest. Cancer Res 2008; 68: 6181–6189.
Gajate C, Barasoain I, Andreu JM, Mollinedo F . Induction of apoptosis in leukemic cells by the reversible microtubule-disrupting agent 2-methoxy-5-(2′,3′,4′-trimethoxyphenyl)-2,4,6-cycloheptatrien-1-one: protection by Bcl-2 and Bcl-X(L) and cell cycle arrest. Cancer Res 2000; 60: 2651–2659.
Andreu JM, Perez-Ramirez B, Gorbunoff MJ, Ayala D, Timasheff SN . Role of the colchicine ring A and its methoxy groups in the binding to tubulin and microtubule inhibition. Biochemistry 1998; 37: 8356–8368.
Abdullah NM, Rosania GR, Shedden K . Selective targeting of tumorigenic cancer cell lines by microtubule inhibitors. PLoS One 2009; 4: e4470.
Rowinsky EK, Donehower RC . Paclitaxel (taxol). N Engl J Med 1995; 332: 1004–1014.
Bonfoco E, Ceccatelli S, Manzo L, Nicotera P . Colchicine induces apoptosis in cerebellar granule cells. Exp Cell Res 1995; 218: 189–200.
Martin SJ, Cotter TG . Disruption of microtubules induces an endogenous suicide pathway in human leukaemia HL-60 cells. Cell Tissue Kinet 1990; 23: 545–559.
Green LS, Donoso JA, Heller-Bettinger IE, Samson FE . Axonal transport disturbances in the vincristine-induced peripheral neuropathy. Trans Am Neurol Assoc 1975; 100: 195–196.
Kuncl RW, Duncan G, Watson D, Alderson K, Rogawski MA, Peper M . Colchicine myopathy and neuropathy. N Engl J Med 1987; 316: 1562–1568.
Zemer D, Revach M, Pras M, Modan B, Schor S, Sohar E et al. A controlled trial of colchicine in preventing attacks of familial Mediterranean fever. N Engl J Med 1974; 291: 932–934.
Ben-Zvi I, Danilesko I, Yahalom G, Kukuy O, Rahamimov R, Liveneh A et al. Risk factors for amyloidosis and impact of kidney transplantation on the course of familial Mediterranean fever. Isr Med Assoc J 2012; 14: 221–224.
Lee SJ, Namkoong S, Ha KS, Nam WD, Kwon YG, Lee H et al. Colchicine-derived compound CT20126 promotes skin allograft survival by regulating the balance of Th1 and Th2 cytokine production. Exp Mol Med 2007; 39: 230–238.
Lee SJ, Nam WD, Na HJ, Cho YL, Ha KS, Hwang JY et al. CT20126, a novel immunosuppressant, prevents collagen-induced arthritis through the down-regulation of inflammatory gene expression by inhibiting NF-kappaB activation. Biochem Pharmacol 2008; 76: 79–90.
Chang JS, Kim SK, Kwon TK, Bae SS, Min DS, Lee YH et al. Pleckstrin homology domains of phospholipase C-gamma1 directly interact with beta-tubulin for activation of phospholipase C-gamma1 and reciprocal modulation of beta-tubulin function in microtubule assembly. J Biol Chem 2005; 280: 6897–6905.
Oh JH, Lee JT, Yang ES, Chang JS, Lee DS, Kim SH et al. The coffee diterpene Kahweol induces apoptosis in human leukemia U937 cells through down-regulation of Akt phosphorylation and activation of JNK. Apoptosis 2009; 14: 1378–1386.
Piperno G, LeDizet M, Chang XJ . Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol 1987; 104: 289–302.
Westermann S, Weber K . Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol 2003; 4: 938–947.
Tokési N, Lehotzky A, Horváth I, Szabó B, Oláh J, Lau P et al. TPPP/p25 promotes tubulin acetylation by inhibiting histone deacetylase 6. J Biol Chem 2010; 285: 17896–17906.
Valenzuela-Fernández A, Cabrero JR, Serrador JM, Sánchez-Madrid F . HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol 2008; 18: 291–297.
Bae SS, Perry DK, Oh YS, Choi JH, Galadari SH, Ghayur T et al. Proteolytic cleavage of phospholipase C-gamma1 during apoptosis in Molt-4 cells. FASEB J 2000; 14: 1083–1092.
Andreu JM, Timasheff SN . Tubulin bound to colchicine forms polymers different from microtubules. Proc Natl Acad Sci USA 1982; 79: 6753–6756.
de Ines C, Leynadier D, Barasoain I, Peyrot V, Garcia P, Briand C et al. Inhibition of microtubules and cell cycle arrest by a new 1-deaza-7,8-dihydropteridine antitumor drug, CI 980, and by its chiral isomer, NSC 613863. Cancer Res 1994; 54: 75–84.
Iliev AI, Djannatian JR, Opazo F, Gerber J, Nau R, Mitchell TJ et al. Rapid microtubule bundling and stabilization by the Streptococcus pneumoniae neurotoxin pneumolysin in a cholesterol-dependent, non-lytic and Src-kinase dependent manner inhibits intracellular trafficking. Mol Microbiol 2009; 71: 461–477.
LeDizet M, Piperno G . Cytoplasmic microtubules containing acetylated alpha-tubulin in Chlamydomonas reinhardtii: spatial arrangement and properties. J Cell Biol 1986; 103: 13–22.
Blagosklonny MV, Robey R, Sackett DL, Du L, Traganos F, Darzynkiewicz Z et al. Histone deacetylase inhibitors all induce p21 but differentially cause tubulin acetylation, mitotic arrest, and cytotoxicity. Mol Cancer Ther 2002; 1: 937–941.
Medina V, Edmonds B, Young GP, James R, Appleton S, Zalewski PD . Induction of caspase-3 protease activity and apoptosis by butyrate and trichostatin A (inhibitors of histone deacetylase): dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer Res 1997; 57: 3697–3707.
Qu X, Du J, Zhang C, Fu W, Xi H, Zou J et al. Arsenic trioxide exerts antimyeloma effects by inhibiting activity in the cytoplasmic substrates of histone deacetylase 6. PLoS One 2012; 7: e32215.
Yang H, Ganguly A, Cabral F . Inhibition of cell migration and cell division correlates with distinct effects of microtubule inhibiting drugs. J Biol Chem 2010; 285: 32242–32250.
Jordan MA, Wilson L . Microtubules as a target for anticancer drugs. Nature Rev Cancer 2004; 4: 253–265.
Decker P, Muller S . Modulating poly (ADP-ribose) polymerase activity: potential for the prevention and therapy of pathogenic situations involving DNA damage and oxidative stress. Curr Pharm Biotechnol 2002; 3: 275–283.
Rosen A, Casciola-Rosen L . Macromolecular substrates for the ICE-like proteases during apoptosis. J Biol Chem 1997; 64: 50–54.
Acknowledgements
This work was supported by a Daejin University grant.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Kim, SK., Cho, SM., Kim, H. et al. The colchicine derivative CT20126 shows a novel microtubule-modulating activity with apoptosis. Exp Mol Med 45, e19 (2013). https://doi.org/10.1038/emm.2013.38
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2013.38
Keywords
This article is cited by
-
C(7)-Derivatives of colchicine with guanosine and biphenyl moieties: molecular modeling, synthesis, and tubulin clustering effect in cancer cells
Russian Chemical Bulletin (2020)
-
Synthesis, Antiproliferative Activity, and Effect on Carcinoma A549 Cell Microtubules of New Tubuloclustin Analogs
Pharmaceutical Chemistry Journal (2019)
