
Abstract
Pelizaeus–Merzbacher disease (PMD) is a rare Mendelian disorder characterised by central nervous system hypomyelination. PMD typically manifests in infancy or early childhood and is caused by mutations in proteolipid protein-1 (PLP1). However, variants in several other genes including gap junction protein gamma 2 (GJC2) can also cause a similar phenotype and are referred to PMD-like disease (PMLD). Whole-exome sequencing in two siblings presenting with clinical symptoms of PMD revealed a homozygous variant in the arginyl-tRNA synthetase (RARS) gene: NM_002887.3: c.[5A>G] p.(Asp2Gly). Subsequent screening of a PMD cohort without a genetic diagnosis identified an unrelated individual with novel compound heterozygous variants including a missense variant c.[1367C>T] p.(Ser456Leu) and a de novo deletion c.[1846_1847delTA] p.(Tyr616Leufs*6). Protein levels of RARS and the multi-tRNA synthetase complex into which it assembles were found to be significantly reduced by 80 and 90% by western blotting and Blue native-PAGE respectively using patient fibroblast extracts. As RARS is involved in protein synthesis whereby it attaches arginine to its cognate tRNA, patient cells were studied to determine their ability to proliferate with limiting amounts of this essential amino acid. Patient fibroblasts cultured in medium with limited arginine at 30 °C and 40 °C, showed a significant decrease in fibroblast proliferation (P<0.001) compared to control cells, suggestive of inefficiency of protein synthesis in the patient cells. Our functional studies provide further evidence that RARS is a PMD-causing gene.
Similar content being viewed by others

Introduction
Pelizaeus–Merzbacher disease (PMD; 312080) is typically an X-linked recessive hypomyelinating leukodystrophy-1 (HLD1) caused by variants in the proteolipid protein-1 gene (PLP1), which is located on chromosome Xq22 and encodes a myelin protein.1, 2 PMD is characterised by nystagmus, hypotonia, motor, and cognitive impairment, severe spasticity, ataxia, and respiratory distress.3, 4 The prevalence of PMD is estimated to be 1 in 300 000 to 500 000 males in the United States.5 Compatible with the X-linked recessive pattern of PMD, heterozygote females have been reported to present a milder phenotype compared to males.6, 7
In addition to PLP1 variants, which are the most common cause of PMD, there is increasing evidence showing that a small fraction of undiagnosed PMD may be caused by variants in other genes including GJC2 (OMIM 608803), SLC16A2 (OMIM 300095), DARS (OMIM 603084), RARS (OMIM 107820), AIMP1 (OMIM 603605), POLR3A (OMIM 614258), POLR3B (OMIM 614366), POLR1C (OMIM 610060), HSPD1 (OMIM 118190), FAM126A (OMIM 610531), TUBB4A (OMIM 602662), VPS11 (OMIM 608549), PYCR2 (OMIM 616406), all of which cause hypomyelination or Pelizaeus–Merzbacher-like disease (PMLD) with clinical manifestations similar to X-linked PMD.8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 Functional comparisons of the genes discussed above are summarised in Table 1.
RARS encodes cytoplasmic arginyl-tRNA synthetase, a member of the aminoacyl-tRNA synthetase (ARS) family of enzymes that are essential for protein synthesis, through specific ligation of each amino acid to its cognate tRNA. RARS protein is a monomeric enzyme and belongs to class I of the aminoacyl-tRNA synthetases.22 It contains well-identified catalytic residues within so-called motifs including His-Ile-Gly-His and LysMet-Ser-Lys (KMSK), which are directly implicated in the chemistry of the aminoacylation reaction.23
Of note, among the ARS proteins, nine are specific for esterification of tRNA with the amino acids Arg, Asp, Gln, Glu, Ile, Leu, Lys, Met, and Pro. These ARS proteins along with three ARS-interacting multifunctional proteins namely p18, p38, and p43, are organised into a macromolecular protein complex called multi-synthetase complex (MSC).24 The MSC, has an estimated molecular weight of 1.5 MDa and is involved in protein synthesis and diverse cellular signalling pathways.25, 26
There are two isoforms of cytoplasmic RARS in human cells. One form is ∼72 kDa, with an N-terminal leucine-zipper domain required for its interaction with AIMP1 in the MSC. The second form is the short ∼60 kDa N-terminal truncated form that is free in the cytoplasm and has no interaction with the MSC.27, 28, 29 Kyriacou and Deutscher have shown that the 72 kDa form is essential for protein synthesis and cell growth in Chinese hamster ovary (CHO) cells, even when a functional 60 kDa RARS is present. Both the 72 and 60 kDa isoforms arise from a single mRNA through the utilisation of alternative AUG initiation codons, displaying similar catalytic properties, and contributing about equally to the total rate of RARS in vitro activity.30
In this study, we performed whole-exome sequencing (WES) in two siblings who presented with suspected PMD and their parents. We identified a homozygous missense variant in RARS in both patients, and upon subsequent screening of a PMD cohort without a genetic diagnosis, we identified compound heterozygous RARS variants in another patient. Functional studies in one of the sibling’s fibroblasts revealed reduced protein levels of RARS and the MSC, and reduced growth rates of patient fibroblasts in arginine depleted media at non-permissive temperatures. In this study, we have provided evidence that the reported variants result in a dysfunctional RARS protein which disrupts the assembly of the MSC, and are the likely cause of the PMD phenotype in our patients.
Materials and methods
Patient clinical information
Patient 1
The first child was born at 41 weeks gestation following an uncomplicated pregnancy and delivery to unrelated parents of Maltese background, with no family history of a neurodegenerative process. He was described as an irritable baby in infancy. He had motor and expressive delay: he smiled responsively at 4 months of age, sat unaided at 2 years, walked at 2½ years, and did not start uttering single words until he was 3 years of age, with short phrases at 4 years. He was noted to have bilateral horizontal nystagmus. During childhood moderate cognitive impairment was apparent, requiring support class schooling. At around 6 years of age, he developed an action tremor and head titubation, and his gait became ataxic. He was in a special class for learning difficulties. There was a fairly static course. When last reviewed clinically at 17 years of age, height and weight were normal. He had coarse horizontal nystagmus with normal ocular fundi. General physical examination was normal. Cranial nerve testing was normal. He had normal muscle bulk with slightly reduced power. Deep tendon reflexes were brisk with ankle clonus and extensor plantar reflexes bilaterally. His gait was described as 'spastic ataxic', and he was unable to tandem-walk.
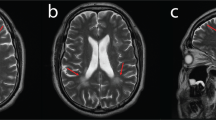
Normal investigations included creatine kinase, blood lactate, thyroid function tests, liver function tests, urine amino and organic acids, oligosaccharides and glycosaminoglycans, plasma very long chain fatty acids, serum transferrin isoforms, biotinidase assay, white blood cell lysosomal enzymes, karyotype, and fragile X testing. A brain MRI scan at 5½ years of age showed marked thinning of the corpus callosum, hypomyelination of the dorsal brainstem, internal capsules and deep white matter, with high T2 signal throughout the periventricular deep white matter. On the fluid-attenuated inversion recovery (FLAIR) sequences used in magnetic resonance imaging, the central white matter was hyperintense when compared to the more peripheral areas of non-myelination, confirming that there appeared to be a dys- or de-myelinating process centrally and non-myelination of the periphery (Figure 1). The findings were suggestive of Pelizaeus–Merzbacher disease. Sequencing of PLP1 only revealed a 3′UTR polymorphism (NM_000533.4 (PLP1_v001): c.*102C>T), and GJC2 sequencing was normal. A repeat MRI scan at 15 years of age showed that the white matter changes had been relatively stable, indicating arrest of myelination that had shown little progression over the years. The white matter volume was markedly reduced within both cerebral hemispheres and accompanied by prominent extra-axial spaces and ex vacuo dilation of the lateral and third ventricles, which had increased slightly in size from the previous scan.

MRI scan of Patient 1 at age 5½ years. (a) Axial T1 IR (inversion recovery) – low signal in white matter consistent with hypomyelination; (b) Axial T2- diffuse white matter hyperintensity consistent with hypomyelination (most important image); (c) Coronal T2 FLAIR_ high signal white matter; (d) Sagittal T1 – thin corpus callosum consistent with white matter volume loss. (e) Axial T1 and (f) axial T2 (inverted contrast) – enhanced cerebrospinal fluid spaces associated with hypomyelination and generalised cortical thinning; (g) Coronal T2 – ventricular dilatation; (h) Sagittal T1 – hypoplasia of the corpus callosum with only the rostral part demonstrable.
Patient 2
The second child, 2 years younger, was also born at term following an uncomplicated pregnancy and delivery. She did not begin walking until 2 years of age and had learning difficulties. She developed nystagmus at around 4 years of age, and her gait was ataxic. She too required support class placement at school. She was also prone to sometimes debilitating anxiety attacks. When last reviewed at age 12½ years, her growth was normal. Her anxiety attacks had resolved without specific therapy. Neuromuscular examination revealed normal tone, normal deep tendon reflexes in the upper limbs but increased in lower limbs. Plantar reflexes were flexor bilaterally. Cerebellar signs were minimal, although she did have dysmetria and dysdiadochokinesia on earlier examinations. She also had truncal ataxia and pendular nystagmus bilaterally. Overall, she was less severely affected than her brother. Her formal ophthalmological assessment confirmed a fine rotatory nystagmus of high frequency and low amplitude, as well as mild astigmatism but normal fundoscopy. Electroretinograms and visual evoked potentials were also normal.
Their mother, whose intellect was normal, was prone to fluttering eye movements with rapid changes in fixation and was found to have multiple small foci of abnormal high signal intensity in the deep white matter on MRI of the brain. Their father and younger brother, now aged 17, have normal general physical and neurological examinations and have had no learning problems.
Patient 3
After an uneventful pregnancy, the male patient was born at term to consanguineous parents (first cousins of Turkish origin). Two aunts and one uncle in the paternal line had suffered from severe incompletely characterised neurological disorders and died between 2 months and 12 years without having learnt to walk or speak. One of these children had also had epilepsy.
The patient first came to medical attention at the age of 3 months because of poor eye fixation and muscular hypotonia. At 10 months of age, he suffered from his first prolonged seizure, followed by intractable focal occipital epilepsy. Additionally, he was prone to startle reactions provoked by noises. He developed microcephaly and severe motor and speech delay: at 14 months of age he had just started to turn from a supine to prone position, was not able to sit and was only able to produce simple noises. Ocular fundi were normal. He developed marked spasticity in the lower limbs. Deep tendon reflexes were brisk with ankle clonus bilaterally and extensor plantar reflexes on the left side. He attended a special class for blind children. When he was last examined at 18 years of age, he was able to sit unaided. He never learned to walk, and could not stand without assistance. General physical examination, as well as height and weight, were normal.
Normal investigations included full blood count, creatine kinase, thyroid function tests, liver function tests, blood ammonia, blood and CSF lactate and pyruvate, plasma amino acids, urine organic acids, and glycosaminoglycans, fibroblast lysosomal enzymes and karyotype. Nerve biopsy revealed multiple discrete distensions in the myelin sheath with disseminated fatty degradation. Muscle biopsy was histologically normal, as was a bone marrow biopsy. Brainstem evoked potentials were delayed. Nerve conduction studies and visual evoked potentials were normal. An EEG was abnormal with diffuse slow rhythms and multifocal epileptiform discharges. Brain MRI was first performed at 1 year of age and repeated at 7 years of age. Both scans revealed dys/hypomyelination of the white matter and hypoplasia of the corpus callosum, with the second MRI scan also revealing generalised cortical thinning, and on sagittal views, only the rostral part of the corpus callosum could be seen. His mother and father and one brother were neurologically normal. DNA from the proband and his parents was available for testing, but cultured skin fibroblasts were not available from the proband for further functional studies.
Whole-exome sequencing
WES performed on P1 and P2 and their parents DNA preparation, WES and bioinformatics analyses were performed as previously described.31
Sanger sequencing
In patients 1 and 2, the RARS variant identified by WES analysis was confirmed by Sanger sequencing of DNA from the siblings and their parents. Subsequently, all 15 exons of the RARS gene were screened in 20 patients with suspected PMD symptoms (provided by BU and Dr Grace Hobson, Alfred I DuPont Hospital for Children, Wilmington, DE, USA) using Sanger sequencing. Parental DNA from patient 3 was also available for Sanger sequencing. Sanger sequencing was performed at the Australian Genome Research Facility, Westmead Millennium Institute (Westmead, Australia), using the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit, and ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Primer sequences and PCR conditions are available on request.
In silico analyses
In silico analyses of the RARS variants were performed using SIFT(http://sift.jcvi.org/), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), GVGD (http://agvgd.iarc.fr/), MutationTaster (www.mutationtaster.org/), PhyloP (http://ccg.vital-it.ch/mga/hg19/phylop), PhastCons (http://compgen.bscb.cornell.edu), Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/), and the HOPE tool (www.cmbi.ru.nl/hope/).
Immunoblotting
For SDS-PAGE, protein was extracted from patient 1 and control fibroblast cells as previously described.32 Thirty micrograms of each protein sample was resolved on a 4–12% Bis-Tris gel (Life Technologies, Carlsbad, CA, USA) and transferred to a polyvinylidene fluoride membrane. Membranes were probed with 1:500 anti-RARS (ab90560 Abcam, Cambridge, UK). Protein loading was normalised to GAPDH (1:2,000 anti-GAPDH for 2 h at room temperature; ab9485, Abcam). All blots were then probed with 1:2500 anti-rabbit (HRP; GE Healthcare, Little Chalfont, Buckinghamshire, UK) for 2 h at room temperature. Membranes were then incubated with enhanced chemiluminescence (ECL) reagents and exposed to Hyperfilm ECL (GE Healthcare). Films were scanned using a SRX-101 A (Konica Minolta Holdings, Inc., Tokyo, Japan) scanner and analysed with ImageJ version 1.49 (http://rsb.info.nih.gov/ij/). Fibroblast pellets from patient 1 were also subjected to Blue Native/PAGE using Native Lysis Buffer (Abcam) and NativePAGE Novex 4-16% Bis-Tris Protein Gels (Life Technologies) according to the manufacturer’s instructions.
Cell culture and growth measurements
For experiments using normal growth medium (containing arginine), fibroblasts were cultured in Dulbecco's modified Eagle medium (DMEM) medium (Life Technologies), supplemented with 10% (v/v) dialysed fetal bovine serum (Bovogen, East Keilor, VIC, Australia), and 100 units/ml penicillin and streptomycin (Life Technologies). For experiments in arginine-free medium, fibroblasts cells were grown in commercial DMEM depleted of arginine and lysine (Life Technologies, A1443101), supplemented with 0.8 mM L-Lysine monohydrochloride (Sigma-Aldrich, L8662) according to the Life Technologies recipe, 10% (v/v) dialysed fetal bovine serum (Bovogen, East Keilor, VIC, Australia), and 100 units/ml penicillin and streptomycin (Life Technologies). All cells were cultured in air containing 5% CO2. Control and patient fibroblasts were seeded in four replicates at 10 × 103 cells/75 cm2 at 30 °C, 37 °C or 40 °C. Cells were collected by trypsinization after 3 days. Cell count and viability were determined in a Muse cell analyser (Merck Millipore, Billerica, MA, USA) using the Muse Count & Viability Reagent (Merck Millipore).
Statistical analyses
Statistical analyses were performed using GraphPad Prism 5.03 (GraphPad Software, San Diego, USA). All variables underwent statistical comparisons by Mann-Whitney U non-parametric testing. A P value less than 0.05 was considered to be statistically significant.
Results
In this study, we identified RARS variants as the cause of PMD in siblings (a male and a female) using WES. Further, an additional 45 samples (from Germany and the USA) were screened for RARS variants where we only identified compound heterozygous variants in one patient from Germany. All three patients presented with nystagmus, motor and expressive delay, moderate cognitive impairment, action tremor and head titubation, and ataxia and spastic gait. There was no history of seizures in patient 1 and 2. The female patient was less severely affected than the male patients, with minimal cerebellar signs and normal electroretinograms. The available MRI findings in two patients were suggestive of PMD.
Since screening of the two most common causative disease genes (PLP1 and GJC2) was normal in the affected siblings, WES was performed. A homozygous c.[5 A>G] p.(Asp2Gly) RARS variant was identified in patient 1 and 2. WES findings were validated by Sanger sequencing, and the parents were found to be heterozygous for the c.[5 A>G] RARS variant (Figure 2a). This variant has been submitted to ClinVar (www.ncbi.nlm.nih.gov/clinvar/variation/162080/).

(a) RARS Sanger sequencing from patients 1, 2 and their parents, showing c.[5 A>G] p.(Asp2Gly) variant is homozygous in both patients and heterozygous in the mother and father. (b, c) Sanger sequencing profile of RARS from patient 3 and parents showing the c.[1367C>T] p. (Ser456Leu) variant is heterozygous in the patient and his mother and the c.[1846_1847delTA] p. (Tyr616Leufs*6) variant is an apparent de novo deletion in the patient (although non-paternity cannot be excluded). (d) Evolutionary sequence conservation of the altered amino acid residue Asp2 is denoted in bold and underlined (e) Evolutionary sequence conservation of the altered amino acid residues Ser456 is denoted in bold and underlined.
Molecular testing in patient 3 for variants (including duplications) in the PLP gene was normal. We subsequently performed Sanger sequencing for all 15 coding exons in the RARS (NM_002887.3) in a cohort of 45 patients with suspected PMD in whom a genetic diagnosis had not been established, including patient 3. We identified compound heterozygous RARS variants c.[1367C>T] and c.[1846_1847delTA] in patient 3 (Figures 2b and c). These variants have been submitted to ClinVar (www.ncbi.nlm.nih.gov/clinvar/variation/265854/ and www.ncbi.nlm.nih.gov/clinvar/variation/265855/ respectively). Sequencing of parental DNA revealed that the mother was heterozygous for the variant c.[1367C>T] whereas the father was variant negative (Figures 2b and c), indicating the patient’s RARS deletion was possibly a de novo event or a consequence of non-paternity.We conducted in silico analyses of the RARS variants for predicted pathogenicity and determination of minor allele frequencies (Table 2). The predicted pathogenicity of the homozygous missense variant [c.5 A>G] p.(Asp2Gly) was variable, with the SIFT and PolyPhen-2 tools predicting it to have no effect, while Mutation-Taster and GVGD predicted it to be damaging. In silico prediction for the c.[1367C>T; p.(Ser456Leu)] variant in patient 3 indicated a deleterious effect by all four predictive software programmes. The c.[1846_1847delTA] p.(Tyr616Leufs*6) variant, which introduces a stop codon at residue 621 of 660 was predicted to be disease causing by Mutation Taster. All three variants were considered rare according to the ExAC with the reported minor allele frequency of c.[5A>G], (a previously reported variant in PMD) being 0.00013 and c.[1367C>T] being 0.00048. There was no reported allele frequency for the c.[1846_1847delTA] variant.
The RARS protein consists of four well-defined structural domains each with specific functions: the ‘N-terminal extension’ domain allows for anchoring of RARS into the MSC; the ‘additional domain’ that allows interaction with the tRNA, made of the D-loop, and T-loop interaction; the‘catalytic domain’ is where the aminoacylation takes place, and the ‘anticodon binding domain’ interacts with the anticodon of the tRNA.33 All three identified variants are found in these functionally characterised domains of the RARS protein; Asp2 is in the N-terminus domain which interacts with the MSC and Ser456 is in the additional domain and is C-terminal to the strictly conserved Arg455 in the KMSKS motif, which stabilises ATP during amino acid activation34 with a possible functional impact, and the p.(Tyr616Leufs*6) variant results in loss of part of the anticodon binding domain.
RARS protein levels were investigated in patient 1 (P1) fibroblasts using immunoblot analysis, revealing a 75% reduction in the 72 kDa RARS isoform in P1 compared to control fibroblasts. Interestingly, the expression levels of the 60 kDa RARS isoform, which lacks the N-terminus, was at a level similar to that of the control fibroblasts (Figure 3a). RARS quantitation could not be performed on the other patients due to unavailability of samples.

(a) Immunoblot showing reduced RARS expression in cultured fibroblasts from patient 1 (P) compared to a control. GAPDH was used as a loading control. (b) BN-PAGE immunoblotting of patient 1 fibroblast (P) and control (C) extracts lysed in 1% digitonin, using antibodies against RARS. GAPDH was used as a loading control.
Since the p.(Asp2Gly) RARS variant lies in the N-terminal domain which interacts with the MSC; we examined assembly of the MSC in patient fibroblasts by Blue Native-PAGE. A 99% reduction in the expression of MSC was observed in fibroblasts from patient 1 (Figure 3b). These results suggest that the p.(Asp2Gly) substitution results in the failure of RARS to be incorporated into the MSC either due to the conformational change or due to lack of total protein expression or both. A previous study has shown that the 72 kDa isoform of RARS is essential for protein synthesis and cell growth in CHO cells despite the presence of the 60 kDa isoform.35 We investigated growth of patient 1 fibroblast’s in arginine depleted media. As shown in Figures 4a and c, at the non-permissive temperatures of 30 °C and 40 °C in arginine depleted media, patient 1 (P1) fibroblast cell growth was reduced by 40% and 30%, respectively, compared with controls fibroblasts (C). However, no difference in cell growth between the control and patient 1 was found at 37 °C (Figure 4b). These observations indicate that patient fibroblasts exhibit temperature-sensitive growth in the absence of the high molecular weight form of RARS.

Patient 1 fibroblasts (P) along with two control fibroblasts (C) were grown in culture medium with and without arginine at (a) 30 °C, (b) 37 °C or (c) 40 °C for 3 days. Viability was determined by counting the number of viable cells using a Muse cell analyser. Data represent the mean of three independent experiments carried out in triplicate.
Using HOPE in silico software, we were able to analyse the structural effects of the amino acid changes (p.(Asp2Gly) and p.(Ser456Leu).36 The wild-type residue (Aspartic acid at position 2) was larger, negatively charged and less hydrophobic compared to the variant amino acid (glycine), which is a neutral amino acid. The altered residue was therefore predicted to be less likely to interact with the MSC.In patient 3, the first variant residue at position 456 (leucine) was larger and more hydrophobic (www.cmbi.ru.nl/hope/). These amino acid changes are predicted to reduce the activity of the RARS protein. Further, calculations on the 3D coordinate of the RARS protein and its sequence annotations was performed using RasMol modelling software (Figure 5).37 Both Asp2 and Ser456 residues are in conserved residues of the RARS protein in higher order species. Interestingly the N-terminus of RARS is not present in some lower species (Figures 2d and e). Unfortunately, it is difficult to use in silico approaches to predict the structural effects of frameshift variants. However, based on the ‘50–55' nucleotides rule, only nonsense variants located more than 50–55 nucleotides upstream of the 3′ most exon-exon junction are likely to be targeted for nonsense mediated decay.38 Thus, as the c.[1846_1847delTA] p.(Tyr616Leufs*6) variant is located less than 50–55 nucleotides upstream of the 3′ most exon-exon junction is not likely to be targeted for nonsense mediated decay. This variant will form a truncated protein, lacking part of the anticodon-binding domain and may be unstable.

3D illustration of the substituted residues Asp2 (black) and Ser456 (yellow) of the RARS protein created with RasMol modelling programme. The RARS domains of N terminal, Catalytic, and anticodon are shown in blue, red, and green respectively.
Discussion
There are numerous reports of the association of tRNA synthetases and many tRNA synthetase cofactors mutations with human diseases, and of these DARS (OMIM 615281), AARS2 (OMIM 615889), DARS2 (OMIM 611105), EARS2 (OMIM 614924), LARS2 (OMIM 615300), MARS2 (OMIM 611390), RARS2 (OMIM 611524), CARS2 (OMIM 612800), FARS2 (OMIM 614946), and AIMP1 (OMIM 260600) affect the central nervous system or brain white matter causing leukodystrophies or leukoencephalopathies.39 Note that most of these genes encode mitochondrial aminoacyl-tRNA synthetases (ARS2).40 Previously reported phenotypes associated with RARS variants are summarised in Table 3.
In addition, variants within RARS (OMIM 616140) have been recently associated with hypomyelination in four patients with identical MRI changes as seen in classic X-linked recessive hypomyelinative leukodystrophy-1, who presented with stagnated motor development, rapid pendular nystagmus, mild intention tremor and dysmetria, ataxia, dysarthria, severe spasticity of legs, and mild mental retardation.15 Our patients demonstrated similar features of motor and expressive delay, bilateral horizontal nystagmus, an action tremor, head titubation, an ataxic gait and moderate to severe cognitive impairment.
We identified a homozygous c.[5 A>G] transition in RARS in patients 1 and 2, resulting in a p.(Asp2Gly) substitution and compound heterozygous variants c.[1367C>T] and c.[1846_1847delTA] in patient 3, resulting in a p.(Ser456Leu) substitution and a truncated protein p.(Tyr616Leufs*6), respectively, with the latter variant being novel. Interestingly, three patients including two sisters and an unrelated individual reported by Wolf et al. (2014) have shared the same variant c.[5 A>G] p. (Asp2Gly), suggesting that the variant can lead to a spectrum of human neurological disease phenotypes (see Table 2 for comparison of phenotypes between the former and current studies). However, it is not clear whether compound heterozygosity versus homozygosity for the c.[5 A>G] variant may result in fundamental differences in disease severity. Further, as there is no evidence of consanguinity in the family history, the prediction of this variant as the cause of hypomyelination can be inferred from the following two possibilities. First, there is the possibility of mere randomness of regions of homozygosity in all populations.41 Second, there is the possibility of remotely shared ancestry.42
The p.(Asp2Gly) substitution in patients 1 and 2 is located in a 72-amino acid domain essential for integration of RARS into the MSC.27 A model of the human RARS protein with the p.(Asp2Gly) variant (Figure 5) suggests a possible loss of internal interactions of the variant residue with the surrounding molecules or domains, with possible loss of external interactions of RARS with the MSC caused by changes in physicochemical properties (http://www.cmbi.ru.nl/hope/). The in silico predictions are in agreement with the immunoblotting and fibroblast growth results showing significant reductions in the 72 kDa RARS isoform and MSC levels in patient 1, and diminished growth potential of the affected cells containing only a single form of RARS at non-permissive temperatures of 30 °C and 40 °C. Mutation in the 72 kDa RARS isoform has been shown to exhibit temperature-sensitive growth characteristics in CHO cells.35 In patient 3, however, as only DNA and no tissue samples or cultured fibroblasts were available to us for functional studies, we had rely on Sanger sequencing, in silico prediction of disease causing for both (c.[1367C>T] and c.[1846_1847delTA]) variants and the phenotypic similarity between patient 3 and the other two patients to support the pathogenicity of the variants in patient 3.
Although the mechanism by which mutations in ARS genes lead to diseases remains obscure, in many cases there is increasing evidence that impairment of aminoacylation activity contributes to the development of neuropathy, emphasising the distinct roles of aminoacylation in neuronal function.43, 44 Thus, establishing an approach for characterising variant alleles in all human ARS-encoding genes will be critical to understand ARS-related neurodegeneration.
In conclusion, our findings suggest that in the absence of a clear PMD diagnosis with mutations in PLP1, screening of the RARS gene could be included in investigations in an appropriate clinical setting. Given our patients’ clinical phenotypes, the location of each of the variants in RARS, and our in silico analysis and functional data, we postulate that the clinical phenotype of our patients can be explained by RARS variants identified in this study.
References
Lossos A, Elazar N, Lerer I et al: Myelin-associated glycoprotein gene mutation causes Pelizaeus-Merzbacher disease-like disorder. Brain 2015; 138: 2521–2536.
Brender T, Wallerstein D, Sum J, Wallerstein R : Unusual presentation of Pelizaeus-Merzbacher disease: female patient with deletion of the proteolipid protein 1 gene. Case Rep Genet 2015; 2015: 453105.
Golomb MR, Walsh LE, Carvalho KS, Christensen CK, DeMyer WE : Clinical findings in Pelizaeus-Merzbacher disease. J Child Neurol 2004; 19: 328–331.
Sima AA, Pierson CR, Woltjer RL et al: Neuronal loss in Pelizaeus-Merzbacher disease differs in various mutations of the proteolipid protein 1. Acta Neuropathol 2009; 118: 531–539.
Xia J, Wang L : Pelizaeus-Merzbacher disease: molecular diagnosis and therapy. Intractable Rare Dis Res 2013; 2: 103–105.
Hobson GM, Kamholz J PLP1-related disorders;in Pagon RA, Adam MP, Ardinger HH et al(eds):GeneReviews. Seattle: University of Washington, 1993.
Carvalho CM, Bartnik M, Pehlivan D, Fang P, Shen J, Lupski JR : Evidence for disease penetrance relating to CNV size: Pelizaeus-Merzbacher disease and manifesting carriers with a familial 11 Mb duplication at Xq22. Clin Genet 2012; 81: 532–541.
Hobson GM, Garbern JY : Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher-like disease 1, and related hypomyelinating disorders. Semin Neurol 2012; 32: 62–67.
Karimzadeh P, Ahmadabadi F, Aryani O, Houshmand M, Khatami A : New mutation of pelizaeus—merzbacher-like disease; a report from iran. Iranian J Radiol 2014; 11: e6913.
Feinstein M, Markus B, Noyman I et al: Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am J Hum Genet 2010; 87: 820–828.
Magen D, Georgopoulos C, Bross P et al: Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am J Hum Genet 2008; 83: 30–42.
Miyamoto Y, Torii T, Eguchi T, Nakamura K, Tanoue A, Yamauchi J : Hypomyelinating leukodystrophy-associated missense mutant of FAM126A/hyccin/DRCTNNB1A aggregates in the endoplasmic reticulum. J Clin Neurosci 2014; 21: 1033–1039.
Shimojima K, Okumura A, Ikeno M et al: A de novo TUBB4A mutation in a patient with hypomyelination mimicking Pelizaeus-Merzbacher disease. Brain Dev 2015; 37: 281–285.
Wolf NI, Vanderver A, van Spaendonk RM et al: Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 2014; 83: 1898–1905.
Wolf NI, Salomons GS, Rodenburg RJ et al: Mutations in RARS cause hypomyelination. Ann Neurol 2014; 76: 134–139.
Nakayama T, Al-Maawali A, El-Quessny M et al: Mutations in PYCR2, encoding pyrroline-5-carboxylate reductase 2, cause microcephaly and hypomyelination. Am J Hum Genet 2015; 96: 709–719.
Edvardson S, Gerhard F, Jalas C et al: Hypomyelination and developmental delay associated with VPS11 mutation in Ashkenazi-Jewish patients. J Med Genet 2015; 52: 749–753.
Thiffault I, Wolf NI, Forget D et al: Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun 2015; 6: 7623.
Lopez-Espindola D, Morales-Bastos C, Grijota-Martinez C et al: Mutations of the thyroid hormone transporter MCT8 cause prenatal brain damage and persistent hypomyelination. J Clin Endocrinol Metab 2014; 99: E2799–E2804.
Cayami FK, La Piana R, van Spaendonk RM et al: POLR3A and POLR3B mutations in unclassified hypomyelination. Neuropediatrics 2015; 46: 221–228.
Biancheri R, Rossi A, Zara F, Filocamo M : AIMP1/p43 mutation and PMLD. Am J Hum Genet 2011; 88: 391, author reply 393-395.
Eriani G, Delarue M, Poch O, Gangloff J, Moras D : Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 1990; 347: 203–206.
Delarue M, Moras D : The aminoacyl-tRNA synthetase family: modules at work. BioEssays 1993; 15: 675–687.
Rémion A, Khoder‐Agha F, Cornu D, Argentini M, Redeker V, Mirande M : Identification of protein interfaces within the multi-aminoacyl-tRNA synthetase complex: the case of lysyl-tRNA synthetase and the scaffold protein p38. FEBS Open Bio 2016; 6: 696–706.
Li R, Macnamara LM, Leuchter JD, Alexander RW, Cho SS : MD Simulations of tRNA and Aminoacyl-tRNA synthetases: dynamics, folding, binding, and allostery. Intern J Molec Sci 2015; 16: 15872–15902.
Park SJ, Ahn HS, Kim JS, Lee C : Evaluation of Multi-tRNA synthetase complex by multiple reaction monitoring mass spectrometry coupled with size exclusion chromatography. PLoS One 2015; 10: e0142253.
Kim HS, Cha SY, Jo CH, Han A, Hwang KY : The crystal structure of arginyl-tRNA synthetase from Homo sapiens. FEBS Lett 2014; 588: 2328–2334.
Yang F, Ji QQ, Ruan LL, Ye Q, Wang ED : The mRNA of human cytoplasmic arginyl-tRNA synthetase recruits prokaryotic ribosomes independently. J Biol Chem 2014; 289: 20953–20959.
Havrylenko S, Mirande M : Aminoacyl-tRNA synthetase complexes in evolution. Int J Mol Sci 2015; 16: 6571–6594.
Zheng YG, Wei H, Ling C, Xu MG, Wang ED : Two forms of human cytoplasmic arginyl-tRNA synthetase produced from two translation initiations by a single mRNA. Biochemistry 2006; 45: 1338–1344.
Alodaib A, Sobreira N, Gold WA et al: Whole-exome sequencing identifies novel variants in PNPT1 causing oxidative phosphorylation defects and severe multisystem disease. Eur J Hum Genet 2016; 25: 79–84.
Riley LG, Menezes MJ, Rudinger-Thirion J et al: Phenotypic variability and identification of novel YARS2 mutations in YARS2 mitochondrial myopathy, lactic acidosis and sideroblastic anaemia. Orphanet J Rare Dis 2013; 8: 193.
Guo M, Schimmel P, Yang XL : Functional expansion of human tRNA synthetases achieved by structural inventions. FEBS Lett 2010; 584: 434–442.
Pang YLJ, Poruri K, Martinis SA : tRNA synthetase: tRNA Aminoacylation and beyond. Wiley Interdiscip Rev RNA 2014; 5: 461–480.
Kyriacou SV, Deutscher MP : An important role for the multienzyme aminoacyl-tRNA synthetase complex in mammalian translation and cell growth. Mol Cell 2008; 29: 419–427.
Venselaar H, te Beek TAH, Kuipers RKP, Hekkelman ML, Vriend G : Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 2010; 11: 548–548.
Arnez JG, Moras D : Structural and functional considerations of the aminoacylation reaction. Trends Biochem Sci 1997; 22: 211–216.
Popp MW, Maquat LE : Leveraging rules of nonsense-mediated mRNA decay for genome engineering and personalized medicine. Cell 2016; 165: 1319–1322.
Simons C, Griffin LB, Helman G et al: Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am J Hum Genet 2015; 96: 675–681.
Szpisjak L, Zsindely N, Engelhardt JI, Vecsei L, Kovacs GG, Klivenyi P : Novel AARS2 gene mutation producing leukodystrophy: a case report. J Hum Genet 2017; 62: 329–333.
Wang J-C, Ross L, Mahon LW et al: Regions of homozygosity identified by oligonucleotide SNP arrays: evaluating the incidence and clinical utility. Eur J Hum Genet 2015; 23: 663–671.
Wang K, Kim C, Bradfield J et al: Whole-genome DNA/RNA sequencing identifies truncating mutations in RBCK1 in a novel Mendelian disease with neuromuscular and cardiac involvement. Genome Med 2013; 5: 67.
Niehues S, Bussmann J, Steffes G et al: Impaired protein translation in drosophila models for Charcot-Marie-Tooth neuropathy caused by mutant tRNA synthetases. Nat Commun 2015; 6: 7520.
Nangle LA, Zhang W, Xie W, Yang XL, Schimmel P : Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc Natl Acad Sci USA 2007; 104: 11239–11244.
Acknowledgements
We are grateful to the NSW Biochemical Genetics Service for establishing initial fibroblast line cultures, and the Molecular Genetics Department for initial DNA extraction, both located at the Western Sydney Genetics Program, Children’s Hospital at Westmead. We are also grateful to Dr Grace Hobson, Alfred I DuPont Hospital for Children, Wilmington, DE, USA, for provision of DNA samples for Sanger sequencing, none of which were shown to have RARS variations. Finally, we are grateful to Dr Shanti Balasubramaniam (Western Sydney Genetics Program, Children’s Hospital at Westmead) and Dr Sebastian Lunke (Translational Genomics Unit, Victorian Clinical Genetics Services) for acquisition and interpretation of some data relating to patient 1. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients included in the study as approved by the Human Research Ethics Committee of the Sydney Children’s Hospitals Network and the Institutional Review Board of Johns Hopkins Medicine.
Author contributions
MN: design and implementation of structural and functional studies, data analysis and manuscript preparation. WG and LR: contribution to design of structural and functional studies, data analysis and manuscript preparation. NS and CB: performance of whole-exome sequencing, implementation of bioinformatics, data analysis and manuscript preparation. KP: interpretation of MRI images for the Australian patients and manuscript preparation. BU: Initial molecular genetic screening and provision DNA sample for the German patient and manuscript preparation. CW: provision of clinical information for the German patient and manuscript preparation. RO: clinical diagnosis and management of the Australian patients and manuscript preparation. JC: overall oversight of the research project, clinical interface, data analysis and manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Nafisinia, M., Sobreira, N., Riley, L. et al. Mutations in RARS cause a hypomyelination disorder akin to Pelizaeus–Merzbacher disease. Eur J Hum Genet 25, 1134–1141 (2017). https://doi.org/10.1038/ejhg.2017.119
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.119
This article is cited by
-
Four pedigrees with aminoacyl-tRNA synthetase abnormalities
Neurological Sciences (2022)
-
Knockdown of Golgi Stress-Responsive Caspase-2 Ameliorates HLD17-Associated AIMP2 Mutant-Mediated Inhibition of Oligodendroglial Cell Morphological Differentiation
Neurochemical Research (2022)
-
Distinct pathogenic mechanisms of various RARS1 mutations in Pelizaeus-Merzbacher-like disease
Science China Life Sciences (2021)
-
Loss of NARS1 impairs progenitor proliferation in cortical brain organoids and leads to microcephaly
Nature Communications (2020)
-
Aminoacyl-tRNA synthetases as therapeutic targets
Nature Reviews Drug Discovery (2019)
