
Abstract
Foveal hypoplasia, always accompanied by nystagmus, is found as part of the clinical spectrum of various eye disorders such as aniridia, albinism and achromatopsia. However, the molecular basis of isolated autosomal recessive foveal hypoplasia is yet unknown. Individuals of apparently unrelated non consanguineous Israeli families of Jewish Indian (Mumbai) ancestry presented with isolated foveal hypoplasia associated with congenital nystagmus and reduced visual acuity. Genome-wide homozygosity mapping followed by fine mapping defined a 830 Kb disease-associated locus (LOD score 3.5). Whole-exome sequencing identified a single missense mutation in the homozygosity region: c.95T>G, p.(Ile32Ser), in a conserved amino acid within the first predicted transmembrane domain of SLC38A8. The mutation fully segregated with the disease-associated phenotype, demonstrating an ∼10% carrier rate in Mumbai Jews. SLC38A8 encodes a putative sodium-dependent amino-acid/proton antiporter, which we showed to be expressed solely in the eye. Thus, a homozygous SLC38A8 mutation likely underlies isolated foveal hypoplasia.
Similar content being viewed by others


Introduction
The fovea, the most central part of the macula that enables sharp vision, is a continuous ganglion cell layer with a high concentration of cone photoreceptors that lacks several of the inner retinal layers as well as retinal vasculature. Foveal hypoplasia is defined as the lack of foveal depression with continuity of all neurosensory retinal layers in the presumed foveal area.1 It is usually described in association with other ocular disorders, such as aniridia, microphthalmia, albinism and achromatopsia.2 Foveal hypoplasia as an isolated entity is a rare phenomenon, with only few cases reported to date.1, 2 All reported cases of foveal hypoplasia are accompanied by decreased visual acuity and nystagmus.1, 2 In the past decade, optical coherence tomography (OCT) has become the preferred methodology in the detection of foveal hypoplasia.3
Foveal hypoplasia has been described to be associated with mutations in numerous genes causing a spectrum of ocular disorders.2 PAX6 (MIM 607108) mutations have been shown to underlie isolated foveal hypoplasia, foveal hypoplasia and presenile cataract syndrome, as well as foveal hypoplasia with anterior segment anomalies (MIM 136520). Foveal hypoplasia is a typical feature in various types of oculocutaneous and ocular albinism, such as autosomal recessive oculocutaneous albinism type IA (OCA1A (MIM 203100)) and oculocutaneous albinism type IB (OCA1B (MIM 606952)) that are due to TYR (MIM 606933) mutations, and X-linked ocular albinism type I (OA1 (MIM 300500)) that is due to GPR143 (MIM 300808) mutations. Another X-linked foveal hypoplasia syndrome is X-linked Aland Island disease (AIED (MIM 300600)), caused by CACNA1F (MIM 300110) mutations, which includes marked impairment of vision, nystagmus, myopia, astigmatism and protan colorblindness.
To date, isolated foveal hypoplasia has been shown to be caused only as part of the wider clinical phenotypic spectrum of PAX6 (autosomal dominant)4 and X-linked GPR143 mutations.5
patients and methods
Patients
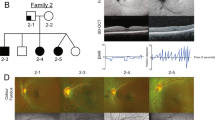
Nine affected individuals (ages 1–65 years) of three apparently unrelated non consanguineous Israeli families of Jewish Indian (Mumbai region) ancestry were studied (Figure 1a). Following Soroka Medical Center IRB approval and informed consent, clinical phenotyping was determined by an experienced ophthalmologist and geneticist for all affected individuals, their parents and siblings. OCT was performed as previously described3 using Spectralis Tracking Laser Tomography (Heidelberg Engineering, Heidelberg, Germany).

Pedigrees, phenotype and mapping: (a) Pedigrees of three affected apparently unrelated non consanguineous Israeli Jewish Mumbai families. A shared homozygosity segment between chr16:83885 and D16S2625 is delineated, Microsatellite markers and their genomic position (in Mb) are presented on the left. (b) Spectral-domain OCT scan of the posterior pole of the right eye of Patient II:3 of pedigree P1 vs normal control individual. As opposed to the control individual (top) with a normal foveal pit (arrow), no foveal structure was demonstrated in the patient (bottom). (c) Autozygosity mapping showing a shared homozygosity locus in chromosome 16 in three affected individuals of pedigree P1 (II:3, II:4 and II:5).
Linkage analysis
Genome-wide linkage analysis was performed using Affymetrix GeneChip Human Mapping 500 K Set Nsp (Affymetrix, Santa Clara, CA, USA) as previously described.6 Homozygosity by descent analysis was carried out using our in-house generated tool for homozygosity mapping.6 Fine mapping and haplotype reconstruction were performed for all available DNA samples of the three pedigrees using polymorphic markers as previously described.7 Microsatellite markers used were as follows: D16S3098, D16S511, D16S534, D16S422, D16S3091, D16S402, chr16:83613, chr16:83885, chr16:84282, D16S2625, D16S3061, D16S3037 and D16S539. All physical positions mentioned are according to the GRCh37/hg19 assembly. Multipoint LOD score for the three pedigrees at the shared locus was calculated using SUPERLINK (http://bioinfo.cs.technion.ac.il/superlinkonline/),8 assuming an autosomal recessive mode of inheritance with penetrance of 0.99 and disease mutant gene frequency of 0.01.
Sequencing
Whole-exome sequencing (HiSeq2000, Illumina, San Diego, CA, USA) was performed using paired-end (2 × 100) protocol at a mean coverage of 30-fold (85–90% of all exonic nucleotides were covered by >10 reads) as previously described.9 For exome enrichment, we used NimbleGen SeqCap EZ Human Exome Library v2.0 (Roche NimbleGen, Madison, WI, USA) targeting 44.1 Mb regions. Sequencing read alignment, variant calling and annotation were performed by DNAnexus (DNAnexus Inc., Mountain View, CA, USA; dnanexus.com).
Restriction analysis
The SLC38A8 c.95T>G mutation abrogates a BtsCI restriction site, enabling differential restriction analysis yielding 105 bp and 145 bp fragments for the wild-type sequence vs a single 250-bp fragment for the mutant variant. PCR primers: Forward- 5′-GGAGGAAAGCGTGAAGACTG-3′; Reverse 5′-GGACTTACCAGCTCCACCAG-3′.
SLC38A8 expression analysis
A panel of cDNA samples extracted (ABgene, Epson, UK) from RNA of various tissues of PBSX1 preperfused C57BL/6 mice was analyzed using two sets of PCR primers designed to amplify cDNA rather than genomic DNA of mouse Slc38a8 and of the beta-2-microglobulin (B2m) housekeeping gene as a control. Primers used for Slc38a8 amplification (320 bp amplicon): Forward 5′-GGCTTTCCTCAGAGTGATCG-3′; Reverse 5′-AGCAGATGGTAGGGAACACG-3′; Primers used for B2m (98 bp amplicon): Forward 5′-TGGTGCTTGTCTCACTGACC-3′; Reverse 5′-TATGTTCGGCTTCCCATTCT-3′.
Results
All patients had nystagmus and subnormal vision since infancy. In two of them, periodic alternating nystagmus with spontaneous alterations of head turn was observed. Patients’ visual acuity ranged between 6/15 and 6/60. Refractive errors were found in all patients and varied between moderate hypermetropia and high myopia. All patients had astigmatism. Six patients had strabismus. All patients had normal cutaneous and ocular pigmentation and no iris transillumination. Fundus exam in all patients revealed foveal hypoplasia. OCT of patient II:3 (pedigree P1) showed the absence of normal foveal pit (Figure 1b). Full-field electroretinography and flash visual evoked potentials testing of six patients were within normal limits. The color vision of four patients as assessed by an Ishihara color vision test was normal. The three patients in pedigree P1 had mild developmental delay and pervasive developmental disorder-like features. One patient in pedigree 3 had diabetes mellitus and hypertension. No other systemic abnormalities were present.
Homozygosity by descent analysis testing all individuals of pedigree P1 identified a 3.4-Mb homozygous segment on chromosome 16q23.3-16q24.1 (Figure 1c) between SNPs rs4888203 and rs6419428, which was common to affected patients of pedigree P1. Fine mapping and haplotype reconstruction, performed for all available DNA samples of the three pedigrees, narrowed the shared homozygosity region to a 830-Kb segment between chr16:83885 and D16S2625, supporting a common ancestral origin for this locus (Figure 1a). Using all the microsatellite markers mentioned above, the three pedigrees combined yielded a maximum LOD score of 3.5 at Chr16:84282.
Whole-exome sequencing data of individual II:3 of pedigree P1 (Figure 1a) were filtered for known variants (dbSNP database and Exome Variant Server, NHLBI Exome Sequencing Project, Seattle,WA, USA; accessed December 2011), revealing no mutations in any of the genes previously associated with isolated foveal hypoplasia (few segments with coverage less than X30 were Sanger sequenced; data not shown). Within the narrow 830-kb locus, only a single mutation was found: c.95T>G missense mutation in exon 1 of SLC38A8 (RefSeq accession number NM_001080442.1), causing a putative p.(Ile32Ser) substitution in the mature protein (Figure 2a). The SLC38A8 mutation (available at http://databases.lovd.nl/shared/genes/SLC38A8), validated by Sanger sequencing, was not reported in the 1000 genomes project (browser.1000genomes.org) and is in a highly conserved amino acid within the putative first transmembrane domain of SLC38A8 (Figures 2b, c). BtsCI restriction analysis demonstrated that the SLC38A8 mutation segregated as expected within the affected families. Of 50 non-related Jews of Mumbai ancestry, 5 were shown to be heterozygous carriers of the mutation. Analysis of SLC38A8 expression in various mouse tissues demonstrated that it is transcribed solely in the eye (Figure 2d).

The SLC38A8 mutation and expression pattern: (a) Sanger sequencing of an unaffected individual (P1, II:2), an obligatory carrier (P1, I:2), and an affected individual (P1, II:3). (b) SMART display of human SLC38A8 transmembrane domain, highlighting the position of the P.(Ile32Ser) mutation. (c) Multiple sequence alignment of SLC38A8 orthologs. The affected isoleucine residue (boxed in red) is conserved across species down to zebrafish. (d) RT-PCR of various normal mouse tissues demonstrating the tissue specificity of Slc38a8.
Discussion
Thorough clinical analysis of our patients demonstrated that the phenotype is limited to isolated foveal hypoplasia, congenital nystagmus and low visual acuity, with variability of the ocular phenotype in different affected individuals. The molecular basis of many cases of isolated nystagmus worldwide is still unknown,10 and the diagnostic modalities used for the diagnosis of foveal hypoplasia before the introduction of OCT could have missed subtle cases.2 Thus, foveal hypoplasia with secondary nystagmus, possibly due to recessive heredity, might be more common than is generally believed.
The inbred Jewish community of the Konkan area of Western India, near Mumbai (formerly Bombay), is often referred to as ‘Bnei Israel’. According to the unwritten folklore of this community, they are either descendants of refugees from ancient Israel (Samaria) after the Assyrian conquest (8th Century BCE) or of a later Jewish migrant group. Some of their ancestors are said to have arrived at the Konkan coast as a consequence of shipwreck in 175 BCE. Of those, tradition says only seven men and seven women survived. After 1750, their descendants moved to Bombay, and in 1947 many of them migrated to Israel. Throughout their known history, Bnei Israel settled in small communities and kept inbred and isolated from other Jews in their vicinity (such as Cochin and Calcutta Jews). In Israel, there still are some small communities with a high prevalence of Bnei Israel descendants.11 Within this extremely inbred small community (estimated 35 000 Bnei Israel Jews in Israel in 2000),11 foveal hypoplasia is common, in line with the high carrier rate we detected.
The only reports identifying the molecular basis of isolated foveal hypoplasia are a single case of a dominant PAX6 missense mutation as part of the aniridia spectrum,4 and a Chinese family with X-linked nystagmus owing to a GPR143 (OA1) mutation,5 where foveal hypoplasia is likely part of the expression of mild ocular albinism commensurate with the expression of GPR143 mainly in pigment cells of the skin and eyes.5 Our patients are distinct in their autosomal recessive inheritance of foveal hypoplasia and lack any signs of oculocutaneous or ocular albinism. The presence of achromatopsia or cone dystrophy was also excluded by normal electroretinogram and color vision. Thus, the anatomical defect of the fovea as evidenced by the OCT seems to be the only cause of their visual handicap and nystagmus. It is presumed that idiopathic infantile nystagmus represents a primary defect in parts of the brain responsible for ocular motor control.2, 10 As SLC38A8 expression is limited to the eye with no expression in the brain, it is likely that the nystagmus caused by the SLC38A8 mutation is secondary to the foveal hypoplasia.
The sodium-coupled neutral amino acid transporters (SNAT) of the SLC38 gene family resemble the classically described System A and System N transport activities in terms of their functional properties and patterns of regulation.12 SLC38 transporters have many physiological roles, including the transfer of glutamine from astrocytes to neurons in the CNS, ammonia detoxification and gluconeogenesis in the liver, and the renal response to acidosis.12 SLC38A8, encoding a putative sodium-dependent amino-acid/proton antiporter, is predicted to have 10 transmembrane domains based on PSORTII, the detection and classification of membrane spanning proteins software (http://psort.hgc.jp/form2.html),13 or 11 such domains per the ‘Kyte Doolittle Hydropathy Plot’ (http://gcat.davidson.edu/DGPB/kd/kyte-doolittle.htm).14 The p.(Ile32Ser) mutation resides within the first predicted transmembrane domain of SLC38A8 (http://smart.embl-heidelberg.de/),15 replacing isoleucine which has a non-polar hydrophobic side chain to serine that has a polar hydrophilic side chain (Figure 2b). Per clustalw2 analysis (http://www.ebi.ac.uk/tools/clustalw2),16 the mutation is at an amino acid that is highly conserved among all vertebrates (Figure 2c). Interestingly, we showed that of 13 different mouse tissues, Slc38a8 was expressed solely in the eye (Figure 2d) and not in other tissues such as the brain (as would be expected in primary nystagmus) or in the skin (as would be expected in albinism-associated phenotypes). In fact, no other gene within the 830-Kb locus is expressed specifically in the eye. The linkage analysis results, put together with the sequencing, restriction analysis and expression data, highly suggest that the phenotype of the patients is due to the SLC38A8 mutation. The precise role of SLC38A8 in normal eye development and function, and the molecular mechanisms through which its mutation likely causes foveal hypoplasia are yet to be elucidated.
Accession codes
References
Mota A, Fonseca S, Carneiro A, Magalhães A, Brandão E, Falcão-Reis F : Isolated foveal hypoplasia: tomographic, angiographic and autofluorescence patterns. Case Rep Ophthalmol Med 2012; 2012: 864958.
Querques G, Prascina F, Iaculli C, Delle Noci N : Isolated foveal hypoplasia. Int Ophthalmol 2009; 29: 271–274.
Holmström G, Eriksson U, Hellgren K, Larsson E : Optical coherence tomography is helpful in the diagnosis of foveal hypoplasia. Acta Ophthalmol 2010; 88: 439–442.
Azuma N, Nishina S, Yanagisawa H, Okuyama T, Yamada M : PAX6 missense mutation in isolated foveal hypoplasia. Nat Genet 1996; 13: 141–142.
Yu Liu J, Ren X, Yang X et al: Identification of a novel GPR143 mutation in a large Chinese family with congenital nystagmus as the most prominent and consistent manifestation. J Hum Genet 2007; 52: 565–570.
Markus B, Birk OS, Geiger D : Integration of SNP genotyping confidence scores in IBD inference. Bioinformatics 2011; 27: 2880–2887.
Feinstein M, Markus B, Noyman I et al: Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am J Hum Genet 2010; 87: 820–828.
Silberstein M, Tzemach A, Dovgolevsky N, Fishelson M, Schuster A, Geiger D : Online system for faster multipoint linkage analysis via parallel execution on thousands of personal computers. Am J Hum Genet 2006; 78: 922–935.
Volodarsky M, Markus B, Cohen I et al: A deletion mutation in TMEM38B associated with autosomal recessive osteogenesis imperfecta. Hum Mutat 2013; 34: 582–586.
Thomas MG, Gottlob I : Optical Coherence Tomography Studies provides new insights into diagnosis and prognosis of infantile nystagmus: A Review. Strabismus 2012; 20: 175–180.
Abbink JG : Ethnic trajectories in Israel comparing the "Bene Israel" and "Beta Israel" Communities, 1950-2000. Anthropos 2002; 97: 3–19.
Mackenzie B, Erickson JD : Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Eur J Physiol 2004; 447: 784–795.
Nakai K, Kanehisa M : A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics 1992; 14: 897–911.
Kyte J, Doolittle RF : A simple method for displaying the hydropathic character of a protein. J Mol Biol 1982; 157: 105–132.
Schultz J, Milpetz F, Bork P, Ponting CP : SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA 1998; 95: 5857–5864.
Larkin MA, Blackshields G, Brown NP et al: Clustal W and Clustal X version 2.0. Bioinformatics 2007; 23: 2947–2948.
Acknowledgements
We thank the Kahn Family Foundation for kind support and the families participating in the study. The study was not funded by the Israeli ministry of health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Perez, Y., Gradstein, L., Flusser, H. et al. Isolated foveal hypoplasia with secondary nystagmus and low vision is associated with a homozygous SLC38A8 mutation. Eur J Hum Genet 22, 703–706 (2014). https://doi.org/10.1038/ejhg.2013.212
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.212
Keywords
This article is cited by
-
Novel compound heterozygous variants of tyrosinase gene in an isolated foveal hypoplasia patient without nystagmus
Journal of Human Genetics (2021)
-
Correlation between electroretinography, foveal anatomy and visual acuity in aniridia due to PAX6 mutations
Documenta Ophthalmologica (2021)
-
Homozygous single nucleotide duplication of SLC38A8 in autosomal recessive foveal hypoplasia: The first Japanese case report
Documenta Ophthalmologica (2021)
-
Autosomal dominant foveal hypoplasia without visible macular abnormalities and PAX6 mutations
Japanese Journal of Ophthalmology (2020)
-
Netzhautverdickung eines 6‑jährigen Jungen
Der Ophthalmologe (2019)
