
Abstract
This paper explores the genetic structure and signatures of natural selection in different sub-populations from the Island of Sardinia, exploiting information from nearly 700 000 autosomal SNPs genotyped with the Affymetrix Genome-Wide Human SNP 6.0 Array. The genetic structure of the Sardinian population and its position within the context of other Mediterranean and European human groups were investigated in depth by comparing our data with publicly available data sets. Principal components and admixture analyses suggest a clustering of the examined samples in two significantly differentiated sub-populations (Ogliastra and Southern Sardinia), as confirmed by AMOVA (FST=0.011; P<0.001). Differentiation of these sub-populations was still evident when they were pooled together with supplementary Sardinian samples from HGDP and compared with several other European, North-African and Near Eastern populations, confirming the uniqueness of the Sardinian genetic background. Moreover, by applying several statistical approaches aimed at assessing differences at the SNP level, the highest differentiated genomic regions between Ogliastra and Southern Sardinia were thus investigated via an extended haplotype homozygosity (EHH)-based test to point out potential selective sweeps. Using this approach, 40 genomic regions were detected, with significant differences between Ogliastra and Southern Sardinia. These regions were subsequently investigated using a long-range haplotype test, which found significant REHH values for SNPs rs11070188 and rs11070192 in the Ogliastra sub-population. In the light of these results and the overlap of the different computed statistics, the region encompassing these loci can be considered a strong candidate to have undergone selective pressure in Ogliastra.
Similar content being viewed by others

Introduction
In recent years, unprecedented advances in the knowledge of human genetic variation have been driven by the technological improvements that have involved genotyping and sequencing platforms, shedding new light on the genetic variability of our species, as well as on the different origins, demographic histories and disease susceptibilities of its populations.1, 2, 3 These advances have also favored a more in-depth understanding of the role that natural selection had in the differentiation processes of human groups after Homo sapiens exited from Africa,4, 5, 6 by enabling researchers to perform studies on the genetic footprints of selection at a higher resolution, with respect to the past, and leading to the identification of hundreds of loci with strong evidence of selection.7, 8, 9, 10, 11, 12, 13 Characterization of these adaptive events clearly represents an extremely precious opportunity to explore the genetic basis of human adaptation and its crucial medical implications, turning out to be considerably important also for the evaluation of the genetic causes behind human diseases.14, 15
Genome-wide scans have been progressively applied at worldwide and regional levels, although most studies have focused primarily on continental European populations.2, 16, 17, 18 The Sardinian genetic variation has begun to be described in the complex landscape of genome-wide investigations, thanks to the high-density genotyping of the HGDP-CEPH panel by Li et al.1 Nevertheless, no studies concerning the island’s internal heterogeneity, or the potential presence of footprints of natural selection in its inhabitants’ genomes, have been reported.
Sardinian populations are undoubtedly of particular interest owing to their genetic background and elevated degree of isolation.19 These characteristics have made them suitable models for studies on monogenic diseases, such as G6PD deficiency,20 Thalassemia21 and Wilson disease.22 Moreover, as the Sardinian population is considered a founder population, the dissection of its genetic variation is also useful for association studies on complex diseases, in particular on autoimmune diseases, such as Type-I Diabetes and Multiple Sclerosis, which are highly represented on the island.23, 24
In order to increase genomic information on Sardinian sub-populations, this research has explored the subregional genetic structure and the signatures of natural selection observable in different samples from the Island of Sardinia. The position of Sardinians in the genetic space of Mediterranean, European and Near-Eastern populations was also investigated. To this end, information on nearly 700 000 autosomal SNPs genotyped with the Affymetrix Genome-Wide Human SNP 6.0 Array (Affymetrix, Inc., Santa Clara, CA, USA) was exploited and used to apply several statistical approaches aimed at assessing differences at the SNP level. The highest differentiated genomic regions between Ogliastra and Southern Sardinia were investigated via an extended haplotype homozygosity (EHH)-based test.5 This demonstrated a potential selective sweep that occurred in the genomes of one of the examined populations.
Materials and methods
Sampling and genotyping
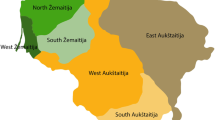
A panel of 321 unrelated healthy individuals, belonging to various regions of the Island of Sardinia (Ogliastra, Trexenta, Sulcis and Campidano), were sampled (Figure 1, Supplementary Table 1S). Individuals were aggregated into two main groups according to their different geographic locations and by taking into account their genetic structure (see Results). The first sub-population includes all samples from Ogliastra, one of the most-isolated regions25, 26, 27, 28 of Sardinia. The second assembles samples from Southern Sardinia (Trexenta, Sulcis and Campidano), a larger and less-isolated area. Population-based methods use these two sub-populations as units of analysis.

Geographical location of the examined Sardinian population samples.
Samples were genotyped with the Affymetrix Genome-Wide Human SNP 6.0 Array. Applying subsequent quality control (QC) filters, a data set made up of 689 879 informative autosomal SNPs with minor allele frequencies >1% was obtained for 310 individuals.
Population structure
Principal component analysis (PCA) was performed on the basis of all informative markers on all the collected Sardinian samples, as well as by exploiting information obtained from a subset of 114 702 SNPs on a data set including literature data from 481 individuals belonging to several European, North-African and Near-Eastern populations (Supplementary Table 2S) and genotyped with different Illumina platforms.
Individual admixture analysis was performed on the two identified sub-populations (Ogliastra and Southern Sardinia) according to a subset of 87 167 SNPs selected in order to have low linkage disequilibrium (LD) values (HR2<0.5) and δ estimates ≥0.05,29 as well as on the data set including literature data on the basis of 92 890 SNPs with HR2<0.5.
An AMOVA test30 between Ogliastra and Southern Sardinia pooled samples was also performed on the basis of 100 000 randomly selected SNPs and 1000 permutations.
LD analysis
LD was measured using the HR2 statistic31 by computing it for Ogliastra and Southern Sardinia and for each couple of SNPs with a physical distance lower than 70.5 kb. For each sub-population, couples of SNPs were clustered in 1-kb-sized windows, which were computed for every 250 base pairs and mean HR2 values were estimated for each window.32
Detection of highly differentiated genomic regions
Highly differentiated genomic regions between the two investigated Sardinian sub-populations were detected by computing three distinct measures of differentiation for each SNP: FST,33 δ and χ2. To merge information from these parameters, we considered the top 35 000 SNPs with highest values (5% of the total SNPs list), filtering for the common markers present in each of the three distributions. From the resulting data set, markers significantly differentiated according to χ2 test were selected. All the identified genomic regions characterized by at least one SNP statistically different between the two Sardinian sub-populations were further investigated in search of signatures of natural selection by applying the long-range haplotype test (LRH).5 Selected genomic regions were phased, and for each identified core SNP, REHH values were calculated. Outliers in the resulting distribution were considered as statistically significant results.
An exhaustive description of the sampling criteria, QC filters and applied statistical analyses is provided in the Supplementary Materials and Methods.
Results
The genetic structures of the population samples examined by means of a PCA were investigated using all the 689 879 informative SNPs that passed QC and data set-filtering procedures. Plots of the top three principal components (PCs) are shown in Supplementary Figure S1. In Supplementary Figure S1A the plots of the first vs second PCs are shown, which represent 4.13% of the whole variability. In particular, the first PC clearly separates Trexenta, Sulcis and Campidano samples (Southern Sardinia) from the Ogliastra region, whereas the second PC underlines a high degree of internal variability in Ogliastra. By plotting the first vs the third PC (3.74% of whole variability), Supplementary Figure S1B confirms the internal differentiation of Ogliastra (in contrast with a lower genetic diversity in Southern Sardinia), as emphasized by the third PC. On the contrary, results from the remaining PCs (data not shown) seem to be not related to geography among and within populations. These data were further compared with the HGDP Sardinian sample,1 exploiting information from the subset of 114 702 SNPs shared between the two different data sets. HGDP individuals from Sardinia seem to be broadly comprised in the variability observed in our previous PCA (Supplementary Figure 2S), in particular showing a higher degree of affinity with Southern Sardinians with respect to individuals from Ogliastra. All Sardinian individuals were further compared with a data set made up of 453 subjects belonging to 26 European, North-African and Near-Eastern ethnic groups (details in Supplementary Table 2S). A plot of first and second PCs (Supplementary Figure 3S), accounting for 15.04% of the total variability, suggests a clear differentiation of the Sardinians with respect to the other examined populations, as well as a considerable internal heterogeneity of the Sardinian population as a whole. In particular, the first PC separates Sardinians from North-African and Near-Eastern populations, whereas the second PC separates them from the Europeans.
With respect to the European groups showing the higher affinity to Sardinians, the French-Basques and the Tuscans seem to be the closest fit according to the first and second PCs, respectively. Sardinians cannot be distinguished from any of the European populations according to the third PC (Supplementary Figure 4S) just as they are inseparable from the Near-Eastern populations on the second PC (Supplementary Figure 3S).
The results of admixture analysis performed on each collected Sardinian sample according to K values ranging from two to five are shown in Supplementary Figure 5S. This plot confirms the results obtained from the PCA study. In particular, at K=2 a clear differentiation between Ogliastra and Southern Sardinia was detectable, whereas for higher K values an internal substructure of the Ogliastra region appeared. This structure indicated three groups of villages (Ulassai with Jerzu, Tertenia with Barisardo and, finally, Villagrande Strisaili), with a distribution reflecting the geographical position of villages in the examined region. At K=5 it was also possible to discern a weak gradient within the Southern Sardinian region, with some differentiation of Sulcis with respect to the other two subregions (Trexenta and Campidano). Admixture analysis was subsequently repeated on the data set, including literature data and exploiting information from a subset of 92 890 SNPs with low LD values (Supplementary Figure 6S). Interestingly, Sardinian individuals, besides maintaining a sharp distinction with respect to samples from other populations, are still characterized by a strong internal subdivision between Ogliastra and Southern Sardinian subjects (in particular at K=5), so that their genetic differentiation results are comparable to those observed among populations from widely larger geographic areas.
The population differentiation between Ogliastra and Southern Sardinia was also evaluated with an AMOVA test based on 100 000 randomly chosen SNPs showing a modest, but statistically significant difference (FST=0.011; P<0.001).
LD, as measured with the HR2 coefficient, is represented as a function of the physical distance between the couples of SNPs shown in Figure 2. As expected, both sub-populations show the well-known LD decay curve, but Ogliastra is characterized by HR2 values higher than those observed for Southern Sardinia for each point of the respective curves.

LD decay for Southern Sardinia (dotted line) and Ogliastra (solid line).
Genomic regions with high differentiation between the two sub-populations have been identified through the calculation of χ2 test, FST and δ values. These measures showed a highly reciprocal correlation (P<10−16), with r=0.978 for FST and χ2, r=0.839 for δ and FST, as well as r=0.881 for δ and χ2. For each of the three obtained distributions, the top 5% SNPs (N=34 394) were considered; then the 22 585 markers shared among the three top groups were retained (Figure 3). After Bonferroni correction for multiple tests, the number of SNPs with significant P-values according to χ2 test was 74. The quality of these markers was checked by means of cluster graphs inspection and four isolated SNPs were removed. The remaining 70 SNPs (Supplementary Table 3S) demarcated different regions, each characterized by a peak SNP with the highest P-values (converted in −log10 scale).

Distribution of FST, δ and −log(P) for χ2 test for all autosomes.
Overall, 40 regions were defined, of which four were characterized by a number of significant markers ranging from four to ten, 1 by three significant SNPs, 8 by two significant SNPs and 27 by a single significant SNP. The 13 regions with at least two relevant SNPs are shown in Table 1.
Each of these regions was phased considering a genomic interval centered on its ‘peak’ SNP, spanning 300 kb upstream and downstream, for a total of 9064 loci. The largest regions were phased equally between the first and the last SNP to achieve a better coverage of the segment under investigation. REHH was calculated for each SNP in both the examined sub-populations and P-values were corrected with the Bonferroni method. In Figure 4, the distributions of REHH values in the bin of allele frequencies are shown. After correction for multiple testing, no significant values resulted for Southern Sardinia as opposed to the Ogliastra region, where REHH values were significant (P=0.01162; REHH=15.86) for two adjacent SNPs (rs11070188, rs11070192). These are located on chromosome 15 at +5 kb from the peak SNP rs7181250, delimited by the C15orf54 and THBS1 genes in 3′ and 5′, respectively, and presenting high FST values (Figure 5). Values of REHH as a function of distance are shown in Figure 6.

Distribution of REHH values in bins of allele frequencies.

FST values in proximity of significant REHH values (rs11070188 and rs11070192).

REHH values for both alleles of rs11070188 as a function of distance. At the top of the figure genes located on the examined region are shown. For rs11070192 the graph does not show detectable differences.
Discussion
In order to increase the genomic and population coverage of past studies on Sardinian populations, the present research explores the subregional geographic structure and signatures of natural selection observable in the genetic variability of distinct sub-populations of the Island of Sardinia belonging to various regions (Ogliastra, Sulcis, Trexenta and Campidano). In addition, this research investigated in depth the genetic position of Sardinians within the context of several other Mediterranean and European human groups. The study exploited information from a data set of 689 879 autosomal SNPs that have been genotyped in 310 Sardinian individuals with the Affymetrix Genome-Wide Human SNP 6.0 Array, as well as from a subset of 114 702 SNPs shared between our experiment and publicly available data sets for several European, North-African and Near-Eastern populations.1, 34
According to the first PC identified in the PCA performed at the single-village level, a clear differentiation between Ogliastra and other samples exists, whereas the other PCs mainly highlight an internal differentiation within Ogliastra; on the contrary, samples from Southern Sardinia appear quite homogeneous. Admixture analysis perfectly confirms these findings, allowing the observation of evident patterns of internal differentiation for the Ogliastra sample. A statistical support for the observed differentiation is also provided by an AMOVA test performed after pooling Southern Sardinian samples together according to PCA and admixture analysis and suggesting a different genetic background for them. Interestingly, population structure analyses carried out at a wider geographical scale, besides confirming the representativeness of HGDP Sardinians for the Island population as a whole, and the high degree of differentiation of Sardinians with respect to other Mediterranean and European populations,1, 35, 36, 37 still highlight an internal differentiation within Sardinia, suggesting that the results of PCA and admixture analysis at the micro-geographical level are not a simple consequence of the adopted small geographical resolution. Moreover, these analyses also point out an appreciable genetic affinity of Sardinians with the French-Basque population that could be explained by their common Paleolithic genetic substrate, which has been much more maintained with respect to other populations as a consequence of their cultural and geographic isolation.38
Differently from our results, some studies that have investigated the Sardinian internal structure pointed out the complete absence of a significant genetic structure by comparing three different macro-areas on the basis of a limited number of autosomal or Y chromosome markers,39, 40 drawing the conclusion that the Sardinian population as a whole is highly homogeneous. A plausible explanation for the difference between literature data and our findings is that results could be influenced by the different surveyed sub-population. Individuals from Ogliastra were not included in previous works and it has been proved that this sub-population is characterized by high genetic isolation and low gene flow.25, 26, 27, 28 These conditions have determined its sharp genetic differentiation with respect to sub-populations belonging to the neighboring areas.25 According to results showing that the pooled sample from Southern Sardinia is quite homogeneous despite being made up of individuals belonging to different historical–geographic subregions, it can be concluded that in Sardinia internal heterogeneity among macro-areas exists, even though it is actually limited to particular areas.
Concerning the internal differentiation in Ogliastra, these results also confirm previous observations, which have detected micro-heterogeneity among small areas or even single villages in the Island of Sardinia including Ogliastra.25, 26, 27, 28, 41, 42, 43, 44, 45 Differences between the two examined sub-populations are also reflected in their LD decay patterns, with Ogliastra showing higher LD values at any distance. This result agrees perfectly with the strong isolation levels reported for Ogliastra, meaning that in this population linkage blocks are averagely longer than those from Southern Sardinia.
To further explore genomic differentiation of the identified sub-populations, three parameters were computed for each of the 689 879 selected informative SNPs, leading to the identification of 40 regions characterized by one or more SNPs statistically differentiated according to χ2 test. In particular, strong levels of differentiation, demonstrated by clusters of SNPs with highly significant P-values after conservative multiple test correction, were detected on chromosomes 1, 9, 12 and 13. Several of these SNPs are located on genes or regions with clinical implications or associated with particular phenotypes in human or animal models;46, 47, 48, 49, 50, 51 therefore, the hypothesis that they could have a role in susceptibility to common complex diseases cannot be ruled out.52 In fact, examination of allele frequencies in 11 HapMap phase III populations has demonstrated high levels of variation for the markers already associated with complex diseases and traits in genome-wide scans.53 It is then plausible that a similar phenomenon could occur also in a limited geographical area such as Sardinia, especially according to the heterogeneity found among its examined groups.
According to this view, the study plan aimed to search for the genomic region showing the highest probability to represent an actual selective sweep. For this purpose, all the differentiated regions mentioned above were phased and analyzed with the LRH test5 for each sub-population. After Bonferroni correction, two SNPs showed significant REHH values in the Ogliastra population: rs11070188 and rs11070192, both mapping on chromosome 15 (15q15) and showing high FST values. It seems worthy to note that rs11070188 is located on a glucocorticoid receptor transcription factor-binding site, and the whole region is delimited by C15orf54 (−81 kb) and THBS1 (+244 kb) genes. The THBS1 gene encodes for Thrombospondin I, a multi-modular secreted protein that associates with the extracellular matrix and exerts a variety of biological functions, such as platelet aggregation, angiogenesis and tumorigenesis. Additionally, the THBS1 gene acts as a receptor for red blood cells parasitized by Plasmodium falciparum, the adhesion of which to small-vessel endothelium enhances Plasmodium virulence. Based on these findings, combined with the overlap of the different computed statistics, the region encompassing these loci can be considered a strong candidate to have undergone a selective pressure in Ogliastra. Although it has been not detected as a target for positive selection in HGDP populations, Sardinians included, by the extensive genome-wide scan carried out by Pickrell et al,7 this research cannot demonstrate the claim for a selective pressure on this genomic region that is specific to the Ogliastra sub-population, as signatures of selection surrounding the THBS1 gene have already been found in two distinct studies.8, 9 In particular, Williamson et al8 detected a selective sweep at 21 kb from the THBS1 gene, according to a combined sample (African-American, European-American, Chinese samples) analysis, whereas Simonson et al9 identified a signal of selection in the same region in a Tibetan population. However, in this specific case, and according to the location of the identified SNPs, both regarding the THBS1 gene, it is possible to speculate about a potential selective pressure represented by malaria infection, which has heavily characterized Sardinia until the complete eradication occurred in the middle of the twentieth century.54 Referring to the classic Fermi data,55, 56 the mean malaria morbidity for Ogliastra, calculated as a weighted average based on the number of individuals for each village, is 57.95%, with respect to the 28.56% for Southern Sardinia (Supplementary Table 4S). These data suggest not negligible malaria endemicity in Ogliastra. The hypothesis of its potential selective pressure on the genomes of individuals inhabiting this area cannot be ruled out, even though further evaluations are required. Furthermore, the fact that the exploited data about malaria morbidity are referred to the first decades of the twentieth century has to be taken into account, so that they likely underestimate the actual burden of malaria from more ancient times. Nevertheless, these data have already shown their usefulness as a reference for analyses focused on the correlation between malaria and genetics.57, 58, 59
In conclusion, although genome-wide scans in search of signatures of natural selection have already been performed, mostly among populations showing high reciprocal divergence levels, the same analyses carried out at the micro-geographical level are still lacking and this research can be considered as a first attempt to partially fill this gap.
The main results highlight an internal differentiation of the Sardinian population, mainly due to a different genetic background of Ogliastra individuals with respect to those from the other examined regions. At the same time, they point out a pattern of homogeneity among samples belonging to regions from Southern Sardinia (Sulcis, Trexenta, Campidano), an area characterized by a considerably lower degree of isolation with respect to Ogliastra.
Future studies based on high-density SNPs genotyping experiments from other Sardinian regions (that is, Northern Sardinia) will undoubtedly contribute to a better knowledge of the internal genetic structure of this island.
The results also support the identification of a potential selective sweep that occurred at the micro-geographical level and concerning the Ogliastra sub-population, albeit more extensive analyses on other Sardinian regions could be useful to strengthen this finding and to carefully identify an underlying biological interpretation.
The highly differentiated genomic regions detected may represent potential targets of the action of natural selection or may present substantial functional differences. It is therefore possible to speculate, on the basis of the high differentiation between the two examined samples, that these could have a role in the susceptibility to common complex diseases.52, 53 These results could have a predictive value for incoming association studies focused on multifactorial diseases in the same sub-populations.
References
Li JZ, Absher DM, Tang H et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008; 319: 1100–1104.
Novembre J, Johnson T, Bryc K et al. Genes mirror geography within Europe. Nature 2008; 456: 98–101.
Xing J, Watkins WS, Witherspoon DJ et al. Fine-scaled human genetic structure revealed by SNP microarrays. Genome Res 2009; 19: 815–825.
Voight BF, Kudaravalli S, Wen X, Pritchard JK : A map of recent positive selection in the human genome. Plos Biol 2006; 4: e72.
Sabeti PC, Varilly P, Fry B et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007; 449: 913–918.
Kelley JL, Swanson WJ : Positive selection in the human genome: from genome scans to biological significance. Annu Rev Genomics Hum Genet 2008; 9: 143–160.
Pickrell JK, Coop G, Novembre J et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res 2009; 19: 826–837.
Williamson SH, Hubisz MJ, Clark AG et al. Localizing recent adaptive evolution in the human genome. Plos Genet 2007; 3: e90.
Simonson TS, Yang Y, Huff CD et al. Genetic evidence for high-altitude adaptation in Tibet. Science 2010; 329: 72–75.
McEvoy BP, Montgomery GW, McRae AF et al. Geographical structure and differential natural selection among North European populations. Genome Res 2009; 19: 804–814.
Novembre J, Di Rienzo A : Spatial patterns of variation due to natural selection in humans. Nat Rev Genet 2009; 10: 745–755.
Amato R, Pinelli M, Monticelli A et al. Genome-wide scan for signatures of human population differentiation and their relationship with natural selection, functional pathways and diseases. Plos One 2009; 4: e7927.
Bigham A, Bauchet M, Pinto D et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. Plos Genet 2010; 6: e1001116.
Bustamante CD, Fiedel-Alon A, Williamson S : A scan for positively selected genes in the genomes of humans and chimpanzees. Nature 2005; 7062: 1153–1157.
Blekhman R, Man O, Hermann L : Natural selection on genes that underlie human disease susceptibility. Curr Biol 2008; 12: 883–889.
Seldin MF, Shigeta R, Villoslada P et al. European population substructure: clustering of northern and southern populations. Plos Genet 2006; 2: e143.
Bauchet M, McEvoy B, Pearson LN et al. Measuring European population stratification with microarray genotype data. Am J Hum Genet 2007; 80: 948–956.
Nelis M, Esko T, Mägi R et al. Genetic structure of Europeans: a view from the North-East. Am J Hum Genet 2009; 85: 679–691.
Cavalli-Sforza LL, Menozzi P, Piazza A : The History and Geography of Human Genes. Princeton, NJ: Princeton University Press, 1994.
Siniscalco M, Bernini L, Latte B, Motulsky AG : Favism and thalassæmia in sardinia and their relationship to malaria. Nature 1961; 190: 1179–1180.
Rosatelli MC, Dozy A, Faa V et al. Molecular characterization of β-thalassemia in the Sardinian population. Am J Hum Genet 1992; 50: 422–426.
Loudianos G, Dessi V, Lovicu M et al. Molecular characterization of wilson disease in the Sardinian population–evidence of a founder effect. Hum Mutat 1999; 14: 294–303.
Karvonen M, Tuomilehto J, Libman I, La Porte R : A review of the recent epidemiological data on the worldwide incidence of type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1993; 36: 883–892.
Pugliatti M, Rosati G, Carton H et al. The epidemiology of multiple sclerosis in Europe. Eur J Neurol 2006; 13: 700–722.
Cappello N, Rendine S, Griffo R et al. Genetic analysis of Sardinia: I. data on 12 polymorphisms in 21 linguistic domains. Ann Hum Genet 60: 125–141.
Angius A, Melis PM, Morelli L et al. Archival, demographic and genetic studies define a Sardinian sub-isolate as a suitable model for mapping complex traits. Hum Genet 2001; 109: 198–209.
Angius A, Bebbere D, Petretto E et al. Not all isolates are equal: linkage disequilibrium analysis on Xq13.3 reveals different patterns in Sardinian sub-populations. Hum Genet 2002; 111: 9–15.
Fraumene C, Petretto E, Angius A, Pirastu M : Striking differentiation of sub-populations within a genetically homogeneous isolate (Ogliastra) in Sardinia as revealed by mtDNA analysis. Hum Genet 2003; 114: 1–10.
Bryc K, Auton A, Nelson MR et al. Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc Natl Acad Sci USA 2010; 107: 786–791.
Excoffier L, Smouse PE, Quattro JM : Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 1992; 131: 479–491.
Sabatti C, Risch N : Homozygosity and linkage disequilibrium. Genetics 2002; 160: 1707–1719.
Jakobsson M, Scholz SW, Scheet P et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature 2008; 451: 998–1003.
Weir BS, Cockerham CC : Estimating F-statistics for the analysis of population structure. Evolution 1984; 38: 1358–1370.
Behar DM, Yunusbayev B, Metspalu M et al. The genome-wide structure of the Jewish people. Nature 2010; 466: 238–242.
Piazza A, Cappello N, Olivetti E et al. A genetic history of Italy. Ann Hum Genet 1988; 52: 203–213.
Lampis R, Morelli L, De Virgiliis S et al. The distribution of HLA class II haplotypes reveals that the Sardinian population is genetically differentiated from the other Caucasian populations. Tissue Antigens 2000; 56: 515–521.
Calò CM, Melis A, Vona G et al. Sardinian population (Italy): a genetic review. Int J Mod Anthrop 2008; 1: 39–64.
Chikhi L, Nichols RA, Barbujani G, Beaumont MA : Y genetic data support the Neolithic demic diffusion model. Proc Natl Acad Sci USA 2002; 99: 11008–11013.
Lampis R, Morelli L, Congia M et al. The inter-regional distribution of HLA class II haplotypes indicates the suitability of the Sardinian population for case-control association studies in complex diseases. Hum Mol Genet 2000; 9: 2959–2965.
Contu D, Morelli L, Santoni F et al. Y-chromosome based evidence for pre-neolithic origin of the genetically homogeneous but diverse Sardinian population: inference for association scans. Plos One 2008; 3: e1430.
Vona G : The peopling of Sardinia (Italy): history and effects. Int J Anthr 1997; 12: 71–87.
Scozzari R, Cruciani F, Pangrazio A et al. Human Y-chromosome variation in the western Mediterranean area: implications for the peopling of the region. Hum Immunol 2001; 62: 871–884.
Zei G, Lisa A, Fiorani O et al. From surnames to the history of Y chromosomes: the Sardinian population as a paradigm. Eur J Hum Genet 2003; 11: 802–807.
Vona G, Calò CM : History of the Sardinian population as inferred from genetic analysis; In Calò CM, Vona G (eds). Human Genetic Isolates. Trivandrum, Kerala, India: Research Signpost, 2006, pp 1–28.
Pistis G, Piras I, Pirastu N et al. High differentiation among eight villages in a secluded area of Sardinia revealed by genome-wide high density SNPs analysis. Plos One 2009; 4: e4654.
Heid IM, Jackson AU, Randall JC et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet 2010; 42: 949–960.
Gesta S, Bezy O, Mori MA et al. Mesodermal developmental gene Tbx15 impairs adipocyte differentiation and mitochondrial respiration. Proc Natl Acad Sci USA 2011; 108: 2771–2776.
De Braekeleer E, Douet-Guilbert N, Rowe D et al. ABL1 fusion genes in hematological malignancies: a review. Eur J Haematol 2011; 6: 361–371.
Kurosu H, Yamamoto M, Clark JD et al. Suppression of aging in mice by the hormone Klotho. Science 2005; 309: 1829–1833.
Arking DE, Krebsova A, Macek M et al. Association of human aging with a functional variant of klotho. Proc Natl Acad Sci USA 2002; 99: 856–861.
Khor CC, Davila S, Shimizu C et al. Genome-wide linkage and association mapping identify susceptibility alleles in ABCC4 for Kawasaki disease. J Med Genet 2011; 48: 467–472.
Myles S, Davison D, Barrett J et al. Worldwide population differentiation at disease-associated SNPs. BMC Med Genomics 2008; 1: 22.
Adeyemo A, Rotimi C : Genetic variants associated with complex human diseases show wide variation across multiple populations. Public Health Genomics 2010; 13: 72–79.
Tognotti E : La Malaria in Sardegna – Per Una Storia Del Paludismo Nel Mezzogiorno (1880–1950). Milano: Franco Angeli, 1996.
Fermi C : Le Regioni Malariche. Decadenza e Risanamento E Spesa ‘Sardegna’ Vol I: Roma: Tipografia editrice di Roma S.A, 1934.
Fermi C : Provincia Di Nuoro. Malaria, Danni Economici, Risanamento E Proposte Per Il Suo Risorgimento Vol II: Sassari: Stamperia della libreria italiana e straniera, 1938.
Gloria-Bottini F, Saccucci P, Magrini A et al. Is there a role of ACP1-ADA1 genetic complex in immune reaction? Association with T1D and with past malarial morbidity. Am J Med Sci 2010; 340: 268–270.
Contu L, Carcassi C, Orrù S et al. HLA-B35 frequency variations correlate with malaria infection in Sardinia. Tissue Antigens 1998; 52: 452–461.
Bottini N, Stefanini L, Lucarelli P et al. Adaptation to past malarial endemia and susceptibility to common diseases in modern populations: a study of adenosine deaminase and MN blood group genetic polymorphisms. Am J Phys Anthropol 2005; 128: 194–198.
Acknowledgements
This work was supported by 4e – ICTSIAI101 (RAS). We thank Cristina and Adam Robinson Marras for language revision of the manuscript. Finally, we thank the anonymous reviewer for constructive comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Piras, I., De Montis, A., Calò, C. et al. Genome-wide scan with nearly 700 000 SNPs in two Sardinian sub-populations suggests some regions as candidate targets for positive selection. Eur J Hum Genet 20, 1155–1161 (2012). https://doi.org/10.1038/ejhg.2012.65
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.65
Keywords
This article is cited by
-
Overcoming the dichotomy between open and isolated populations using genomic data from a large European dataset
Scientific Reports (2017)
-
Complex interplay between neutral and adaptive evolution shaped differential genomic background and disease susceptibility along the Italian peninsula
Scientific Reports (2016)
-
Geographic population structure analysis of worldwide human populations infers their biogeographical origins
Nature Communications (2014)

{kind=link}
{kind=link}