
Abstract
Autism spectrum disorders (ASDs) are a heterogeneous group of neurodevelopmental disorders, including childhood autism, atypical autism, and Asperger syndrome, with an estimated prevalence of 1.0–2.5% in the general population. ASDs have a complex multifactorial etiology, with genetic causes being recognized in only 10–20% of cases. Recently, copy-number variants (CNVs) have been shown to contribute to over 10% of ASD cases. We have applied a custom-designed oligonucleotide array comparative genomic hybridization with an exonic coverage of over 1700 genes, including 221 genes known to cause autism and autism candidate genes, in a cohort of 145 patients with ASDs. The patients were classified according to ICD-10 standards and the Childhood Autism Rating Scale protocol into three groups consisting of 45 individuals with and 69 individuals without developmental delay/intellectual disability (DD/ID), and 31 patients, in whom DD/ID could not be excluded. In 12 patients, we have identified 16 copy-number changes, eight (5.5%) of which likely contribute to ASDs. In addition to known recurrent CNVs such as deletions 15q11.2 (BP1-BP2) and 3q13.31 (including DRD3 and ZBTB20), and duplications 15q13.3 and 16p13.11, our analysis revealed two novel genes clinically relevant for ASDs: ARHGAP24 (4q21.23q21.3) and SLC16A7 (12q14.1). Our results further confirm the diagnostic importance of array CGH in detection of CNVs in patients with ASDs and demonstrate that CNVs are an important cause of ASDs as a heterogeneous condition with a variety of contributory genes.
Similar content being viewed by others


Introduction
Autism spectrum disorders (ASDs) (OMIM 209850) are a heterogeneous group of neurodevelopmental diseases characterized by impairments in social interactions, communication deficits, stereotyped behaviors, and a restricted range of interests.1, 2, 3, 4 Twin studies have shown a strong genetic contribution to the etiology of ASDs;1, 2 however, ASDs are considered to have a complex multifactorial etiology, involving several chromosomal regions. The search for genes associated with autism is far from complete.
The genetic etiology of ASDs is currently recognized only in ∼10–20% of cases, including single-gene disorders such as fragile X syndrome (FMR1, OMIM 300624), tuberous sclerosis (TSC1 and TSC2, OMIM 191100, 613254), Rett syndrome (MECP2, OMIM 312750), PTEN macrocephaly syndrome (OMIM 605309), Timothy syndrome (CACNA1C, OMIM 601005), and Joubert syndrome (AHI1, OMIM 608629), as well as chromosomal aberrations such as maternally derived duplication 15q11q13 (OMIM 608636) and deletion 22q13.3 (SHANK3, OMIM 606232).2, 4, 5, 6 Other genes that have an important role in neuronal activity that is, NRXN1, NLGN3, NLGN4, and CNTNAP2 have also been shown to be pathogenic for autism.4, 7 Recently, Xu et al8 used a meta-analysis of published data to show that over 400 genes associated with ASDs map to pathways important for neuronal activity, including neuroactive ligand–receptor interaction, synapse transmission, and axon guidance.
Microarray studies from multiple laboratories have shown that copy-number variants (CNVs) contribute to ∼10% of ASDs cases. Jacquemont et al9 have found CNVs in 27.5% of patients with syndromic ASDs, while Sebat et al10 have identified de novo CNVs in 10% of simplex families with autism and in 3% of patients with an affected first-degree relative. Similar results have been obtained by Marshall et al,11 who identified de novo CNVs in ∼7% of patients with sporadic ASDs and in ∼2% of families with affected siblings. In contrast, Pinto et al4 have reported de novo CNVs in 5.6 and 5.5% of simplex and multiplex families, respectively. Results of studies by Sanders et al12 and Levy et al13 on a large number of simplex families have confirmed the significance of rare de novo events for idiopathic autism. Christian et al14 have reported CNVs in 11.6% of patients with ASDs (14% de novo and 86% inherited). In addition, Bremer et al15 have reported clinically significant CNVs in 8% of patients with ASDs, of which 4% were de novo events and 9% of CNVs were of unknown clinical significance. Qiao et al16 have studied 100 patients with ASDs and described nine pathogenic known and novel CNVs. Glessner et al3 have found CNVs enriched with genes encoding neuronal cell-adhesion molecules (NLGN1 and ASTN2) and involved in the ubiquitin pathways (UBE3A, PARK2, RFWD2, and FBXO40). Cusco et al2 have identified copy-number changes in 12.5% of patients with idiopathic ASDs and implicated the phosphatidylinositol signaling pathway and the glutamatergic synapse in the etiology of the disorder.
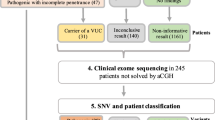
We have used genome-wide exon-targeted array CGH to identify CNVs in a cohort of 145 patients with ASDs with (45) or without (69) developmental delay/intellectual disability (DD/ID) and 31 patients in whom DD/ID could not be excluded. In 12 patients, we have identified 16 non-polymorphic copy-number changes, including three clinically relevant novel rare de novo CNVs and five potentially pathogenic changes important for ASDs.
Patients and methods
Patients
This is the first CNV study in patients with ASDs of the Polish ethnicity. We have studied 145 subjects (123 males and 22 females, ranging in age from 3–26 years) with ASDs, including autism, Asperger syndrome, and pervasive developmental disorder with atypical autism from 132 simplex and 13 multiplex families. Forty-five patients had additional DD/ID. The disorders were classified according to the ICD-10 standards and the Childhood Autism Rating Scale (CARS) protocol. All boys have tested negative for Fragile X syndrome. Informed consents approved by the institutional review board, for Bioethics Commission at the Institute of Mother and Child, were obtained in all cases.
The following three patients have de novo CNVs that we believe are pathogenic for ASDs and patient 4 has two inherited potentially pathogenic CNVs.
Patient 1 is a 6-year-old boy diagnosed with atypical autism (ICD-10: F 84.1), and severe ID (ICD-10: F 72). He was delivered spontaneously without complications at 38 weeks of gestation to a mother with one previous pregnancy and delivery. Apgar scores were 5, 6, and 7 points at 1, 5, and 10 min of life, respectively. Body parameters were in normal range with birth weight 2950 g and head circumference 34 cm. Ultrasound showed a CNS hemorrhage and agenesis of the corpus callosum. Hypothyroidism and abnormal CNS myelination pattern were also documented during early childhood. Psychomotor development was significantly delayed from birth, but he was able to sit unsupported at 12 months of age and walk at 23 months of age. Patient presented with hypotonia with protracted shoulders and kyphosis. He was asthenic with poorly developed subcutaneous tissue and uncoordinated and irregular movements with significantly reduced gross and fine motor skills. Currently, he walks independently but needs help in climbing stairs, requires assistance in dressing and undressing, and eats using his hands. He has a generalized significant impairment of cognitive functions and presents limited understanding of simple orders. He demonstrates poor interest in surroundings, responds to simple verbal instructions in well-known situations, and exhibits delayed reaction times. He is able to recognize objects, colors, and sort objects according to simple characteristics. He has poor communication skills defined by poor eye contact, expressionless face, lack of expressive speech, rarely producing any sounds, and expressing basic needs non-verbally, using body language signals for physiological needs. Occasionally, he produces single sounds like ‘ah’ and ‘oh’ to express emotional states. Behaviorally, he is not very active, has a low energy level, demonstrates a significantly reduced ability to imitate and cooperate in tasks, and presents with stereotyped movements and autostimulation.
Patient 2 is a 7-year-old girl diagnosed with autism (ICD-10: F 84.0) and was the product of the first pregnancy. Her mother had gestational edema and suffered from chronic sinusitis. The proband was delivered spontaneously at 41 weeks of gestation with normal body parameters: weight 4000 g, length 57 cm, and head circumference 36 cm. Her Apgar scores were 9 at 1 and 5 min of life. She was able to sit independently at 8 months and walked unassisted at 13 months. At ∼14 months, regression of skills involving social contact was apparent: she stopped imitating, her movements became stereotypical, and she did not develop verbal communication. Currently, she is physically fit but not very active with slightly reduced motor coordination, manual dexterity, and graphomotor skills although she prefers motor activity. She presents with generalized impairment of cognitive functions and limited understanding of basic commands; although she is able to learn simple activities, she has difficulty in generalizing these experiences. A strong point in her development is visual perception and she is able to recognize objects, colors and shapes, and can sort objects correctly based on these features. She presents with disorders of social interaction, and lacks expressive speech, produces inarticulate sounds, and expresses basic needs non-verbally using body language and gestures. Her ability to imitate is limited and she exhibits numerous repetitive behaviors. Her ability to initiate social contacts is reduced, although she cooperates in simple tasks when encouraged, but often eats inedible objects such as crayons. She has limited level of functioning in daily activities, but is independent in basic self-care tasks. She had an abnormal pattern of EEG at central and parietal CNS regions.
Patient 3 is a 10-year-old boy diagnosed with autism (ICD-10: F 84.0) and mild ID (ICD-10: F70). He was delivered spontaneously at 41 weeks of gestation with an Apgar score of 10 points at 1 and 5 min and normal body parameters, with weight 3600 g and length 55 cm. Motor development reached normal milestones until ∼2 years of age, when he began to regress in skills involving social contact, exhibited increasingly stereotypical activities, and failed to develop verbal communication until after 4 years of age. Currently, he is physically fit and active, with a tendency toward hyperactivity. He has protracted shoulders, a flat thorax, with slightly reduced manual dexterity, and graphomotor skills. He is overly sensitive to environmental stimulation, exhibiting a particularly strong reaction in response to touch and auditory stimuli. He presents increased emotional excitability and lability with little tolerance to change. He communicates needs and can describe his surroundings verbally using single words and simple sentences, but has a poor vocabulary, shows incorrect articulation and does not maintain conversations. There are many instances of immediate and delayed echolalia and his understanding of verbal cues is limited to learned activities and daily tasks. A strong point in his development is visual perception. He has the ability to recognize and differentiate between objects and colors, correctly creating simple categories for objects based on their features. He is able to read, write, work on simple tasks, and solve simple mathematical problems of addition and subtraction, with the ability to use decimals properly.
Patient 4 is a 12-year-old boy diagnosed with autism (ICD-10: F 84.0), epilepsy (ICD-10: G 40), and mild ID (ICD-10: F 70). He was delivered spontaneously at 38 weeks of gestation as the product of his mother’s first pregnancy, which was complicated by uterine infection. Apgar scores were 10 at 1 and 5 min of life. Body parameters were in the normal range: weight 3050 g, head circumference 33 cm, and length 50 cm. He had pneumonia in the 1st month of life with subsequent frequent upper respiratory infections. He had colicky pains until the 5th month of life. After reaching the age of 2 years, he suffered from pneumonia (probably caused by an allergy) 2–3 times per month. Early childhood cognitive and social activities were disordered and autostimulation was observed. His active speech was severely delayed and gradually developed after the 4th year of life. At 9 years of age, he began showing symptoms of precocious puberty and was treated by an endocrinologist. He was diagnosed with epilepsy at the age of 10 years. Currently, he presents with scoliosis, asymmetric posture, muscle hypotonia, slightly reduced motor coordination, and graphomotor skills. He communicates verbally with a reduced level of expressional speech and comprehension. His cognitive development is reduced and incoherent. Complications in concentration are observed. His behavior presents typical schemes for diagnosed disorders. He cooperates in play and school/task situations, communicates with simple sentences, understands complex tasks, and gradually builds answers to open questions. He understands incoherencies in the pictures, consequences of actions that are familiar, and adds and subtracts numbers over 10. He does not cooperate with abstract tasks, and has poor understanding of complex social situations. His brother was also diagnosed with autism (F 84.0) and his father’s brother has schizophrenia. His mother is healthy but her family history is positive for ID (two cousins).
DNA isolation
Genomic DNA extracted from peripheral blood cells using a Puregene DNA Blood Kit (Qiagen, Gentra System, Minneapolis, MN, USA) according to the manufacturer’s protocol was used for array-CGH and Fragile X syndrome analysis.
Chromosomal microarray analysis
Custom-designed oligonucleotide array CGH was performed using clinical exon-targeted chromosomal microarray analysis V8.0 and V8.1 OLIGO designed by Medical Genetics Laboratories at Baylor College of Medicine (BCM) (http://www.bcm.edu/geneticlabs/?pmid=16207) in cooperation with Department of Medical Genetics at Institute of Mother and Child and manufactured by Agilent Technology (SantaClara, CA, USA). This array contains the ‘best-performing’ oligonucleotides, selected from Agilent’s library and has been further empirically optimized. Genomic features of the V8.0 and V8.1 OLIGO design include interrogation of all known microdeletion and microduplication syndrome regions, as well as pericentromeric, subtelomeric, and computationally predicted NAHR-mediated genomic instability regions flanked by low-copy repeats and has additional exon coverage for over 1700 known and candidate genes for neurodevelopmental disorders. The entire nuclear genome is covered with an average resolution of 13 kb, excluding low-copy repeats and other repetitive sequences.17 Digestion, labeling, and hybridization were performed, following the manufacturer’s protocols. The BCM web-based software platform and a custom-designed IMiD-web2py software were used for evaluating the significance of the genomic copy-number changes. The genomic coordinates are based on the February 2009 assembly of the reference human genome (GRCh37/hg19). In our analyses, we consider at least three consecutive oligos, thus the actual resolution of the platform is ∼40 kb. On average, each patient had seven CNVs that we interpreted as the polymorphic changes. To classify the identified variants, we have considered their type (deletion or duplication), size, gene content, inheritance pattern, and available information from the BCM and public CNV databases and followed the guidelines of the American College of Medical Genetics.
Fluorescence in situ hybridization (FISH) analysis
FISH analysis was used to verify aberrations identified by array CGH studies and for investigating parental samples. Metaphase chromosomes and interphase nuclei were extracted from phytohemagglutinin-stimulated peripheral blood lymphocytes, according to the standard procedures. Parental origin of the identified CNVs was studied using FISH with the same probes or array CGH.
Results
We have identified 16 non-polymorphic CNVs, ranging in size from 140 kb to 4.5 Mb in 12 patients studied (patients 4, 7, and 8 had more than one non-polymorphic CNV). We have found CNVs in two individuals (patients 5 and 6) from a cohort of 69 patients with the isolated ASDs. Of 44 patients with ASDs and DD/ID, six subjects have CNVs (patients 1, 3, 4, 9–11). Moreover, out of 32 patients in whom DD/ID could not be excluded, we have found CNVs in four individuals (patients 2, 7, 8, and 12). We have classified these non-polymorphic CNVs into three groups: clinically relevant, potentially pathogenic, and likely benign for ASDs (Tables 1, 2, 3).
Whereas deletion 15q11.2 (BP1-BP2) in patient 6 and duplications 15q13.3 in patient 4 and 16p13.11 in patient 7 are known common recurrent CNVs, the other changes are unique and nonrecurrent. The deletions in patients 1–3 arose de novo (Table 1), and were not found in a Database of Genomic Variants (http://projects.tcag.ca/variation), Autism Chromosome Rearrangement Database (http://projects.tcag.ca/autism/), or a group of 2026 healthy individuals.18 However, non-paternity was not excluded in families 1–3. CNVs in patients 4, 5, 7–12 were inherited from the apparently healthy parents (Tables 2 and 3). For the remainder of CNVs, the parental samples were not available.
Discussion
Our chromosomal microarray analysis revealed three clinically relevant and five potentially pathogenic CNVs in 7 out of 145 patients with ASDs studied (5.5%). All three de novo CNVs were identified in the simplex families (2.3%). However, our cohort is too small for statistically significant comparisons.
A de novo ∼4.5-Mb deletion in 3q13.2q13.31, encompassing the DRD3 (dopamine receptor D3) and ZBTB20 (zinc finger and BTB domain-containing protein 20) genes, has been identified in a 6-year-old boy with atypical autism and severe DD and ID. Very recently, deletions of this genomic region have been described in 14 patients with DD, postnatal overgrowth, hypoplastic male genitals, and characteristic facial features.19 Eight of these patients presented with autism or attention deficits. The authors proposed DRD3 and ZBTB20 as strong candidate genes for DD.
In patient 2 with autism, speech delay, and abnormal EEG, we have identified a de novo ∼1.0-Mb deletion in 4q21.23q21.3, harboring only one gene ARHGAP24 that encodes a Rho-GTPase-activating protein, regulating neuronal growth. Nguyen et al20 showed that deregulation of ARHGAP24 inhibits axon and dendrite outgrowth and branching. A number of overlapping larger-sized deletions, ranging in size between 4.4 and 17.9 Mb have been found in patients with ASDs and DD9 (case 5; Decipher patients 753, 994, 1599, 4665, and 254502). Thus, we propose ARHGAP24 as a good candidate gene for ASDs.
Very little is known about SLC16A7 (solute carrier family 16, member 7), the only gene removed by a de novo ∼520-kb deletion in 12q14.1, identified in a 10-year-old patient 3 with autism and mild ID. Two Decipher patients (2218 and 2473) with overlapping deletions, ranging in size between 6.0 and 10.1 Mb had autistic behavior and speech delay. Given that SLC16A7 deletion in our patient 3 arose de novo and three other SLC family genes, SLC9A9, SLC6A4, and SLC25A12 are well-established autism genes, we suggest that SLC16A7 is a good candidate gene for ASDs.
In patient 4 with autism, epilepsy, attention deficit, speech delay, mild ID, and truncal hypotonia, we have found an ∼480-kb deletion in 11q24.3q25, removing a single gene SNX19, and a common recurrent small CHRNA7 duplication in 15q13.3 (Figure. 1). Small CHRNA7 duplications have been found in general population in 1/185 individuals.21 ASDs have been reported in 4/11 patients with small CHRNA7 duplications referred for chromosomal microarray analysis; however, its pathogenicity remains controversial.22, 23 Recently, Williams et al24 reported association of the CHRNA7 duplication with attention-deficit hyperactivity disorder. Interestingly, in contrast to patient 4, his brother, who has the SNX19 deletion but not the CHRNA7 duplication, does not present problems with concentration. If causative (with variable expressivity and incomplete penetrance), CHRNA7 duplication could be the most common CNV responsible for ASDs. SNX19 maps to the Jacobsen syndrome (JS, OMIM 147791) critical region25 and is expressed in the brain. Ji et al26 suggested that SNX19, in addition to THYN1, OPCML, NCAPD3, and NTM, may be responsible for DD/ID in patients with Jacobsen syndrome (JS). A number of patients with JS have also neurobehavioral abnormalities, including attention deficit and ASDs. We hypothesize that haploinsufficiency of SNX19 found in patient 4, his affected brother, and their healthy mother may contribute to these features in patients with JS.

(a) Array CGH analysis in patient 4, showing an ∼0.5-Mb deletion in chromosome 11q24.3q25 (red) and duplication in 15q13.3 (green). Orange dots indicate known benign CNVs to serve as a positive control of hybridization. (b) Only one gene, SNX19, is deleted. (c) Results of the FISH analysis with the BAC clone RP11-385B5 (green) and the centromeric probe SE11 Kreatech (aqua) used as a control. White arrow indicates the deleted region. The color reproduction of this figure is availabe at the European Journal of Human genetics online.
Moreover, we have identified a maternally inherited ∼140-kb deletion at 18q21.33q21.33 that contains the PIGN (phosphatidylinositol-glycan, class N) gene. Homozygous mutations in PIGN have been reported in patients with autosomal recessive multiple congenital anomalies–hypotonia–seizures syndrome (MCAHS; OMIM 614080).27
BP1–BP2 deletion in 15q11.2 has been reported in patients with schizophrenia, epilepsy, and ASDs,28, 29 and it could contribute to Asperger syndrome observed in our patient 6.
Hannes et al30 suggested that the recurrent duplication at 16p13.11 may be a benign variant.30 However, other reports associated this CNV with a variety of neuropsychiatric and neurobehavioral disorders, including autism, schizophrenia, intellectual disabilities, cognitive impairment, attention deficit hyperactivity disorder, and epilepsy, as well as congenital heart defects, skeletal manifestations, and thoracic aortic aneurysms and dissections.31, 32, 33, 34, 35, 36, 37 We suggest that this duplication may contribute to the autistic features seen in patient 7.
In summary, our results further confirm the diagnostic importance of array CGH in detection of CNVs in patients with ASDs. We propose that ARHGAP24 and SLC16A7 may be responsible for ASDs.
References
Bucan M, Abrahams BS, Wang K et al: Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet 2009; 5: e1000536.
Cusco I, Medrano A, Gener B et al: Autism-specific copy number variants further implicate the phosphatidylinositol signaling pathway and the glutamatergic synapse in the etiology of the disorder. Hum Mol Genet 2009; 18: 1795–1804.
Glessner JT, Wang K, Cai G et al: Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009; 459: 569–573.
Pinto D, Pagnamenta AT, Klei L et al: Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010; 466: 368–372.
Betancur C : Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res 2011; 1380: 42–77.
Miles JH : Autism spectrum disorders -a genetics review. Genet Med 2011; 13: 278–294.
Robinet C, Pellerin L : Brain-derived neurotrophic factor enhances the expression of the monocarboxylate transporter through translational activation in mouse cultured cortical neurons. J Cereb Blood Flow Metab 2010; 30: 286–298.
Xu LM, Li JR, Huang Y, Zhao M, Tang X, Wei L : AutismKB: an evidence-based knowledgebase of autism genetics. Nucleic Acids Res 2012; 40: D1016–D1022.
Jacquemont ML, Sanlaville D, Redon R et al: Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet 2006; 43: 843–849.
Sebat J, Lakshmi B, Malhotra D et al: Strong association of de novo copy number mutations with autism. Science 2007; 316: 445–449.
Marshall CR, Noor A, Vincent JB et al: Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 2008; 82: 477–488.
Sanders SJ, Ercan-Sencicek AG, Hus V et al: Multiple recurrent de novo CNV, including duplications of the 7q11.23 Williams Syndrome region, are strongly associated with autism. Neuron 2011; 70: 863–885.
Levy D, Ronemus M, Yamrom B et al: Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 2011; 70: 886–897.
Christian SL, Brune CW, Sudi J et al: Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol Psychiatry 2008; 63: 1111–1117.
Bremer A, Giacobini M, Eriksson M et al: Copy number variation characteristics in subpopulations of patients with autism spectrum disorders. Am J Med Genet B Neuropsychiatr Genet 2011; 156: 115–124.
Qiao Y, Riendeau N, Koochek M et al: Phenomic determinants of genomic variation in autism spectrum disorders. J Med Genet 2009; 46: 680–688.
Boone PM, Bacino CA, Shaw CA et al: Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat 2010; 31: 1326–1342.
Shaikh TH, Gai X, Perin JC et al: High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res 2009; 19: 1682–1690.
Molin AM, Andrieux J, Koolen DA et al: A novel microdeletion syndrome at 3q13.31 characterised by developmental delay, postnatal overgrowth, hypoplastic male genitals, and characteristic facial features. J Med Genet 2012; 49: 104–109.
Nguyen LS, Jolly L, Shoubridge C et al: Transcriptome profiling of UPF3B/NMD-deficient lymphoblastoid cells from patients with various forms of intellectual disability. Mol Psychiatry 2011; e-pub ahead of print 20 December 2011; doi:10.1038/mp.2011.163.
Sharp AJ, Mefford HC, Li K et al: A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet 2008; 40: 322–328.
Szafranski P, Schaaf CP, Person RE et al: Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: benign or pathological? Hum Mutat 2010; 31: 840–850.
Cooper GM, Coe BP, Girirajan S et al: A copy number variation morbidity map of developmental delay. Nat Genet 2011; 43: 838–846.
Williams NM, Franke B, Mick E et al: Genome-wide analysis of copy number variants in attention deficit hyperactivity disorder: the role of rare variants and duplications at 15q13.3. Am J Psychiatry 2012; 169: 195–204.
Bernaciak J, Szczałuba K, Derwińska K et al: Clinical and molecular-cytogenetic evaluation of a family with partial Jacobsen syndrome without thrombocytopenia caused by an approximately 5 Mb deletion del(11)(q24.3). Am J Med Genet A 2008; 146A: 2449–2454.
Ji T, Wu Y, Wang H, Wang J, Jiang Y : Diagnosis and fine mapping of a deletion in distal 11q in two Chinese patients with developmental delay. J Hum Genet 2010; 55: 486–489.
Maydan G, Noyman I, Har-Zahav A et al: Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 2011; 48: 383–389.
de Kovel CG, Trucks H, Helbig I et al: Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain 2010; 133: 23–32.
Sahoo T, Theisen A, Rosenfeld JA et al: Copy number variants of schizophrenia susceptibility loci are associated with a spectrum of speech and developmental delays and behavior problems. Genet Med 2011; 13: 868–880.
Hannes FD, Sharp AJ, Mefford HC et al: Recurrent reciprocal deletions and duplications of 16p13.11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J Med Genet 2009; 46: 223–232.
Ullmann R, Turner G, Kirchhoff M et al: Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat 2007; 28: 674–682.
Behjati F, Shafaghati Y, Firouzabadi SG et al: M-banding characterization of a 16p11.2p13.1 tandem duplication in a child with autism, neurodevelopmental delay and dysmorphism. Eur J Med Genet 2008; 51: 608–614.
Kirov G, Grozeva D, Norton N et al: Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet 2009; 18: 1497–1503.
Williams NM, Zaharieva I, Martin A et al: Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet 2010; 376: 1401–1408.
Kuang SQ, Guo DC, Prakash SK et al: Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet 2011; 7: e1002118.
Nagamani SC, Erez A, Bader P et al: Phenotypic manifestations of copy number variation in chromosome 16p13.11. Eur J Hum Genet 2011; 19: 280–286.
Ramalingam A, Zhou XG, Fiedler SD et al: 16p13.11 duplication is a risk factor for a wide spectrum of neuropsychiatric disorders. J Hum Genet 2011; 56: 541–544.
Acknowledgements
We are grateful to the patients and to their families for participation in this study. We thank Linda J Guynn for helpful discussion. We thank Drs BR Brinkley, AL Beaudet, and JR Lupski for facilitating the collaboration between the Institute of Mother and Child and Baylor College of Medicine. The work was supported by grant R13-0005-04/2008 from the Polish Ministry of Science and Higher Education. Magdalena Bartnik is supported by the START fellowship from the Foundation for Polish Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Wiśniowiecka-Kowalnik, B., Kastory-Bronowska, M., Bartnik, M. et al. Application of custom-designed oligonucleotide array CGH in 145 patients with autistic spectrum disorders. Eur J Hum Genet 21, 620–625 (2013). https://doi.org/10.1038/ejhg.2012.219
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.219
Keywords
This article is cited by
-
Analysis of a Protein Network Related to Copy Number Variations in Autism Spectrum Disorder
Journal of Molecular Neuroscience (2019)
-
Identification of novel candidate disease genes from de novo exonic copy number variants
Genome Medicine (2017)
-
A customized high-resolution array-comparative genomic hybridization to explore copy number variations in Parkinson’s disease
neurogenetics (2016)
-
3p22.1p21.31 microdeletion identifies CCK as Asperger syndrome candidate gene and shows the way for therapeutic strategies in chromosome imbalances
Molecular Cytogenetics (2015)
-
Array-CGH in children with mild intellectual disability: a population-based study
European Journal of Pediatrics (2015)
