
Abstract
Wagner syndrome (WS) is an autosomal dominant vitreoretinopathy affecting various ocular features and is caused by mutations in the canonical splice sites of the VCAN gene, which encodes the large chondroitin sulfate proteoglycan, versican. We report the identification of novel splice acceptor and donor-site mutations (c.4004−1G>C and c.9265+2T>A) in two large WS families from France and the United Kingdom. To characterize their pathogenic mechanisms we performed qRT-PCR experiments on RNA from patient-derived tissues (venous blood and skin fibroblasts). We also analyzed RNA from the original Swiss family reported by Wagner (who has the previously reported c.9265+1G>A mutation). All three mutations resulted in a quantitative increase of transcript variants lacking exons 7 and/or 8. However, the magnitude of the increase varied between tissues and mutations. We discuss altered balance of VCAN splice variants in combination with reduction in glycosaminoglycan protein modifications as possible pathogenic mechanisms.
Similar content being viewed by others

Introduction
Wagner syndrome (WS; OMIM143200) is a rare, dominantly inherited eye disorder belonging to the group of VCAN-related vitreoretinopathies (reviewed in Kloeckener-Gruissem and Amstutz1). The vitreous of affected individuals show the hallmark features of an optically empty vitreous with strands, membranes and/or veils. The symptoms of WS frequently manifest during adolescence but can start in early childhood. The most vision-threatening feature of WS is retinal detachment1, 2 (Nas et al, 2012, personal communication). Other features of WS include myopia, pre-senile cataract, progressive night blindness and chorioretinal atrophy. Systemic abnormalities have not been described. The genetic cause of WS is mutation of the VCAN gene (formerly called CSPG2). VCAN has 15 exons and encodes versican, a large chondroitin sulfate proteoglycan of 3396 amino acids. Versican is an extracellular matrix (ECM) protein and a component of the vitreous, where it is likely to be involved in maintenance and structural integrity. Four transcript/protein isoforms of versican are known: V0 (NM004385.4), V1 (NM001164097), V2 (NM001164098) and V3 (NM001126336). These variants result from the alternative splicing of exons 7 and 8. V0 contains both exons, V1 lacks exon 7, V2 lacks exon 8 and V3 lacks both exon 7 and 8. All four variants are found in many tissues including the eye.3, 4 Glycosaminoglycan (GAG) residues are post-translationally attached to those protein domains encoded by exons 7 and 8. It is thought that these residues participate in formation of the vitreous gel.5 To date, all WS patients, for whom mutations have been identified, have been heterozygous for single-nucleotide substitutions in either the conserved splice acceptor site of intron 7 or the splice donor site of intron 8.3, 6, 7, 8, 9, 10 Although the exact molecular pathogenesis of WS is not fully understood, it is clear that splicing is involved. Mutations in the intron 7 splice acceptor site can activate a cryptic splice site 21 base pairs into exon 8 generating aberrant transcripts.3, 8, 10 Small amounts of aberrant transcripts have also been found in blood of WS patients carrying the c.9265+1G>A mutation.6 In addition to aberrant transcripts, intron 7 splice acceptor-site mutations significantly increase the amount of the normal splice variants V2 and V3 in patients’ lymphoblastoid cells.3, 8 We investigated whether a similar pattern of disturbed alternative splicing is seen in other families with WS-causing VCAN splice-site mutations. We identified the VCAN mutations in two families with WS and analyzed transcript isoforms V2 and V3 in fibroblasts and peripheral blood from members of these families, and a third family with a previously described VCAN mutation. Our results suggest that V2/V3 overexpression is indeed a shared feature of VCAN splice-site mutations but that tissue- and mutation-specific factors influence the extent of altered splicing.
Material and methods
Subjects
French family from Lyon (FL) and British family from Cardiff (BC) (Figure 1): phenotypes in these families and their linkage to chromosome 5q14.3 have previously been described.11, 12, 13 Genomic DNA samples from 38 family FL members (19 with WS) and from three affected family BC members (IV3, IV10 and V4) were subjected to Sanger sequencing of the intron 7/exon 8 and exon 8/intron 8 boundaries of VCAN. Primers, designed using Primer3 (vs 0.4.0) (http://frodo.wi.mit.edu/primer3/), are listed (Supplementary Table S1). Polymerase HotFire (Solis Biodyne, Tartu, Estonia) was used for standard PCR; a skin biopsy and venous blood for RNA (PreAnalytiX PAXgene tubes (Qiagen, Hilden, Germany)) was obtained from affected female III/13 (family FL) and from the index patient of the original Wagner family (OW).6 Fibroblast cell cultures were established.14 Fibroblasts from the BC family were not available but venous blood for RNA from patient IV3 was available.

Pedigrees of studied families. (a) French family from Lyon: all numbered individuals, except generation I, and individuals II4 and IV3 were subjected to DNA analysis. Skin biopsy was taken from III13 (arrow) for fibroblast cultures and venous blood was collected for transcript analysis. (b) British family from Cardiff: *individuals IV3, IV10 and V4 were subjected to DNA analysis. Individual IV3 had venous blood collected for RNA analysis. Full black symbol indicates Wagner syndrome; half full symbol indicates blindness due to unknown cause.
Transcript analysis
For in silico analysis the following prediction programs were used: Human Splicing Finder (HSF) version 2.4 (http://www.umd.be/HSF/); the MaxEntScan algorithm (part of the HSF interface); NNSplice (http://www.fruitfly.org/seq_tools/splice.html); NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) and ESEfinder: (http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi).15
For quantitative transcript analysis RNA was isolated using an RNeasy mini kit (fibroblasts) and Qiagen PAX-preparation kit (blood) (Qiagen). Quality and quantity of RNA was assessed using Agilent 2100 Bioanalyzer on micro RNA chips (Agilent Technology AG, Basel, Switzerland). cDNA was synthesized using reverse transcriptase III and random hexamers for priming (Invitrogen, Basel, Switzerland). Quantitative transcript analysis (qRT-PCR) was performed by measuring the incorporation of SYBR-green (http://www.appliedbiosystems.com). Transcript isoform-specific primers (Supplementary Table S1; Figure 2b) were optimized to yield 90–110% efficiency and to apply ΔΔCt method for calculation of relative expression.16 qRT-PCR of 18S rRNA served as endogenous control. In a 384-well plate 10 μl reaction volume contained 1 μl 10 × PCR buffer with 1.5 mM MgCl2, 1 μl 2 mM dNTP, 0.08 μl Taq Polymerase, 0.25 μl 10 × SYBR-green (including ROX) and 2 ng cDNA for VCAN transcript analysis or 2 pg for 18S rRNA. Concentration of primers was determined to be: 1.34 μ M for V0, 1.10 μ M for V1, 1.25 μ M for V2, 1.28 μ M for V3, and 2.5 μ M for 18S rRNA. The ABI 7900 HT Sequence detection system was programmed for initial 2 min at 50 °C and 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C, 45 s at 60 °C and 1 min at 72 °C. Dissociation curves were established. Each qRT-PCR measurement was repeated between 5 and 15 times. Fibroblast/blood RNA from unaffected unrelated individuals served as controls, to which all patient samples were compared.

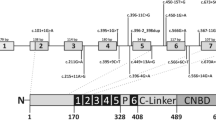
Electropherogram of mutations and diagram of VCAN gene and alternative splice variants. (a) Electropherogram showing mutations at c.4004+1G>C (splice acceptor) in family FL and c.9265+2T>A (splice donor) in family BC. Angled arrows indicate the position of mutations within genomic intronic22 region. Exons 2–6 and 9–15 are fused. Exons 7 and 8 are subject to RNA splicing, which generates splice variants (V0, V1, V2 and V3) displayed in b. Horizontal arrows and numbers above the transcripts indicate the position of primers used for qRT-PCR and the expected size of amplicons in base pair (bp). Oval symbols on protein isoforms, symbolized with N and C terminus, indicate various protein binding sites. H indicates binding of hyaluronin. Black vertical lines on shades parts of the proteins, encoded by exons 7 and 8, indicate attached glucosaminoglycan residues.
Results
DNA sequence analysis
Families FL and BC (Figure 1) segregate dominantly inherited eye disease consistent with WS, and linkage to 5q14.3 though mutations had not previously been identified.11, 12, 13 In 19 affected individuals of the FL family we found a novel heterozygous mutation, c.4004−1G>C, which maps to the conserved splice acceptor site of the intron 7/exon 8 boundary (Figure 2a). It is absent in 19 unaffected individuals from the pedigree. Similarly, in the BC family we found a novel heterozygous mutation, c.9265+2T>A, which maps to the conserved splice donor site of exon 8/intron 8. All three affected individuals tested had the mutation. Both mutations were not found in 300 control alleles.
RNA analysis
At first, we used five prediction programs to analyze the strength of the exon 8 splice sites, comparing the reference sequence with a range of reported and the two novel splice-site mutations in VCAN. The predicted strength was severely reduced by mutations (Table 1). To experimentally support these predictions, quantitative analysis of VCAN transcripts was performed for each of the naturally occurring splice variants (V0, V1, V2 and V3) in fibroblasts from a member of the FL and OW families, and blood from all three families. The amount of splice variants V2 and V3 was significantly increased in all WS patient samples. This increase was quantitatively distinct not only when comparing fibroblasts with blood, but also when comparing different mutations (Figure 3). In fibroblasts, splice donor-site mutation c.9265+1G>A caused a ∼700-fold increase in V2 compared with a ∼80-fold increase caused by c.4004−1G>C. An inter-mutation difference was not observed for the V3 variant in fibroblasts (both showed ∼150-fold increase). In blood, there was elevated V2 levels associated with both c.4004−1G>C (64-fold) and c.9265+1G>A (48-fold) but these were not statistically different from each other. However, the c.9265+2T>A mutation caused significantly greater expression of V2 than the other two mutations (230-fold). The V3 variant was also increased in blood, but the magnitude of the increase varied with mutation (c.4004−1G>C, 6-fold; c.9265+1G>A, 32-fold; and c.9265+2T>A, 143-fold). Variants V0 and V1 did not show statistically significant differences to the control samples in fibroblasts or blood (data not shown).

qRT-PCR analysis of VCAN variants V2 and V3. Relative expression of transcript variants measured from fibroblasts and blood are shown in bar diagram. RNA from control individual was set to 1. The mottled bars in V2 indicates the presence of exon 7. Error bars represent confidence intervals (95% level); technical replicates of 5. Arrows indicate position of primers for qRT-PCR, as shown in Figure 2.
Discussion
We have identified two novel VCAN mutations (c.4004−1G>C and c.9265+2T>A) in families with WS. As with previously reported mutations causing WS 3, 6, 7, 8, 9, 10, 17 they mapped to the conserved splice sites flanking exon 8. Our in silico analyses suggested that WS mutations consistently reduce the strength of the affected splice sites, promoting exon skipping. The observed increase of the V2 and V3 transcript isoforms, lacking exons 8 and 7, may be a consequence. In support, increased V2 expression (∼80-fold) of WS patients with mutations in the splice acceptor site (c.4004−1, −2 and −5) has been indicated previously.3, 8 We are now showing that the magnitude of this increase appears to be modulated by mutation- and tissue-specific factors, a possible explanation of the apparent contradiction between in silico and experimental data.
Within the human eye, transcripts for all four versican isoforms have been found in the retina,3 the trabecular meshwork and the ciliary muscle.4 As eye tissues from WS patients are difficult to access, we studied blood and skin fibroblasts. It remains possible that the observed changes in VCAN expression are not reflected in key eye tissues, such as the vitreous and retina. VCAN is expressed in many tissues,3, 18 yet patients with WS have not been reported to manifest disease outside the eye. The lack of extra-ocular features may be due to differences in VCAN expression between tissues. Alternatively, it may reflect differential susceptibility to pathological consequences.
The vitreous is almost avascular and could be considered a form of ECM as it contains ∼0.1% macromolecules, some of them include hyaluronan and typical ECM proteins such as collagen, proteoglycans and glycoproteins, such as versican, that are embedded in the highly hydrated matrix (98–99% water).5, 19 The general function of the non-collagen elements appears to lie in spacing, binding and linking other ECM components.20 Glucosaminoglycans (GAGs) can fill space, bind and organize water and interfere with negatively charge molecules. Versican, a proteoglycan with chondroitin sulfate modifications, may contribute to these functions, especially through GAGs, which are attached in the domains encoded by exons 7 and 8 (Figure 2b). Isoforms without exon 8 (V2) or exons 7 and 8 (V3) have substantially reduced or no GAG attachment sites, possibly lending them distinct functional properties as described for V3 in muscle tissue.21 Assuming a change in transcript levels is reflected in the balance of protein isoforms, our data support this hypothesis. Alteration of V2 and V3 stochiometry with other macromolecules may contribute to the molecular basis of WS pathology possibly through failure to prevent a collapse of collagen fibrills due to reduction of GAG residues, which then can cause increased density of the vitreous and the formation of strands or veils.
The pathogenesis of WS may also be influenced by aberrant VCAN splice products, as these have been observed in varying amounts in blood and lymphoblastoid cells from patients with mutations in splice donor and acceptor sites.3, 6, 10 Whether such aberrant transcripts are translated and can exert functional effects is not known. Imbalance of the V0 and V1 variants is difficult to assess as neither we nor Mukhopadhyay et al8 could detect statistically significant differences. If there are quantitative differences, they will be of smaller magnitude and therefore harder to measure.
Taken together we suggest that a quantitative imbalance of the normally occurring splice variants is a major contributor to the pathology. Further understanding of the pathogenesis of WS will emerge from the analysis of additional families. Interestingly, not all patients with WS carry mutations in the canonical splice sites of VCAN,8 suggesting some unexplained genetic heterogeneity. When mutations are eventually identified in these patients (in new loci or other regulatory elements of VCAN) this information will give us valuable insights into the pathological processes underlying WS.
References
Kloeckener-Gruissem B, Amstutz C : VCAN-related vitreoretinopathy; in Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (eds): Gene Reviews. Seattle (WA): University of Washington, 2009.
Jansen LM : Retinopathy after nivaquine treatment. Ophthalmologica 1962; 144: 438–444.
Miyamoto T, Inoue H, Sakamoto Y et al: Identification of a novel splice site mutation of the CSPG2 gene in a Japanese family with Wagner syndrome. Invest Ophthalmol Vis Sci 2005; 46: 2726–2735.
Zhao X, Russell P : Versican splice variants in human trabecular meshwork and ciliary muscle. Mol Vis 2005; 11: 603–608.
Theocharis DA, Skandalis SS, Noulas AV et al: Hyaluronan and chondroitin sulfate proteoglycans in the supramolecular organization of the mammalian vitreous body. Connect Tissue Res 2008; 49: 124–128.
Kloeckener-Gruissem B, Bartholdi D, Abdou MT, Zimmermann DR, Berger W : Identification of the genetic defect in the original Wagner syndrome family. Mol Vis 2006; 12: 350–355.
Meredith SP, Richards AJ, Flanagan DW, Scott JD, Poulson AV, Snead MP : Clinical characterisation and molecular analysis of Wagner syndrome. Br J Ophthalmol 2007; 91: 655–659.
Mukhopadhyay A, Nikopoulos K, Maugeri A et al: Erosive vitreoretinopathy and wagner disease are caused by intronic mutations in CSPG2/Versican that result in an imbalance of splice variants. Invest Ophthalmol Vis Sci 2006; 47: 3565–3572.
Ronan SM, Tran-Viet KN, Burner EL, Metlapally R, Toth CA, Young TL : Mutational hot spot potential of a novel base pair mutation of the CSPG2 gene in a family with Wagner syndrome. Arch Ophthalmol 2009; 127: 1511–1519.
Brezin AP, Nedelec B, Barjol A, Rothschild PR, Delpech M, Valleix S : A new VCAN/versican splice acceptor site mutation in a French Wagner family associated with vascular and inflammatory ocular features. Mol Vis 2011; 17: 1669–1678.
Fryer AE, Upadhyaya M, Littler M et al: Exclusion of COL2A1 as a candidate gene in a family with Wagner-Stickler syndrome. J Med Genet 1990; 27: 91–93.
Perveen R, Hart-Holden N, Dixon MJ et al: Refined genetic and physical localization of the Wagner disease (WGN1) locus and the genes CRTL1 and CSPG2 to a 2- to 2.5-cM region of chromosome 5q14.3. Genomics 1999; 57: 219–226.
Zech JC, Morle L, Vincent P et al: Wagner vitreoretinal degeneration with genetic linkage refinement on chromosome 5q13-q14. Graefes Arch Clin Exp Ophthalmol 1999; 237: 387–393.
Glaus E, Schmid F, Da Costa R, Berger W, Neidhardt J : Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR splice defect in patient-derived cells. Mol Ther 2011; 19: 936–941.
Robinson DO, Lin F, Lyon M et al: Systematic screening of FBN1 gene unclassified missense variants for splice abnormalities. Clin Genet 2011; e-pub ahead of print September 6 2011 doi:10.1111/j.1399-0004.2011.01781.x.
Livak KJ, Schmittgen TD : Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25: 402–408.
Brunak S, Engelbrecht J, Knudsen S : Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol 1991; 220: 49–65.
Bode-Lesniewska B, Dours-Zimmermann MT, Odermatt BF, Briner J, Heitz PU, Zimmermann DR : Distribution of the large aggregating proteoglycan versican in adult human tissues. J Histochem Cytochem 1996; 44: 303–312.
Theocharis AD, Papageorgakopoulou N, Feretis E, Theocharis DA : Occurrence and structural characterization of versican-like proteoglycan in human vitreous. Biochimie 2002; 84: 1237–1243.
Ponsioen TL, Hooymans JM, Los LI : Remodelling of the human vitreous and vitreoretinal interface – a dynamic process. Prog Retin Eye Res 2010; 29: 580–595.
Lemire JM, Merrilees MJ, Braun KR, Wight TN : Overexpression of the V3 variant of versican alters arterial smooth muscle cell adhesion, migration, and proliferation in vitro. J Cell Physiol 2002; 190: 38–45.
Lima BL, Santos EJ, Fernandes GR et al: A new mouse model for marfan syndrome presents phenotypic variability associated with the genetic background and overall levels of Fbn1 expression. PLoS One 2010; 5: e14136.
Acknowledgements
We thank the patients for participation. We also thank Esther Glaus, and Mariana Wittmer from the Institute of Medical Molecular Genetics for their excellent support with the DNA sequencing facility.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Kloeckener-Gruissem, B., Neidhardt, J., Magyar, I. et al. Novel VCAN mutations and evidence for unbalanced alternative splicing in the pathogenesis of Wagner syndrome. Eur J Hum Genet 21, 352–356 (2013). https://doi.org/10.1038/ejhg.2012.137
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.137
Keywords
This article is cited by
-
Identification of key genes in atrial fibrillation using bioinformatics analysis
BMC Cardiovascular Disorders (2020)
-
Intravitreal Injection of Splice-switching Oligonucleotides to Manipulate Splicing in Retinal Cells
Molecular Therapy - Nucleic Acids (2015)
