
Abstract
RNA viruses can rapidly mutate and acquire resistance to drugs that directly target viral enzymes, which poses serious problems in a clinical context. Therefore, there is a growing interest in the development of antiviral drugs that target host factors critical for viral replication, since they are unlikely to mutate in response to therapy. We recently demonstrated that phosphatidylinositol-4-kinase IIIβ (PI4KIIIβ) and its product phosphatidylinositol-4-phosphate (PI4P) are essential for replication of enteroviruses, a group of medically important RNA viruses including poliovirus (PV), coxsackievirus, rhinovirus, and enterovirus 71. Here, we show that enviroxime and GW5074 decreased PI4P levels at the Golgi complex by directly inhibiting PI4KIIIβ. Coxsackievirus mutants resistant to these inhibitors harbor single point mutations in the non-structural protein 3A. These 3A mutations did not confer compound-resistance by restoring the activity of PI4KIIIβ in the presence of the compounds. Instead, replication of the mutant viruses no longer depended on PI4KIIIβ, since their replication was insensitive to siRNA-mediated depletion of PI4KIIIβ. The mutant viruses also did not rely on other isoforms of PI4K. Consistently, no high level of PI4P could be detected at the replication sites induced by the mutant viruses in the presence of the compounds. Collectively, these findings indicate that through specific single point mutations in 3A, CVB3 can bypass an essential host factor and lipid for its propagation, which is a new example of RNA viruses acquiring resistance against antiviral compounds, even when they directly target host factors.
Similar content being viewed by others


Introduction
The genus Enterovirus belongs to the family of Picornaviridae, a group of non-enveloped positive-strand RNA viruses that includes many important human and animal pathogens. Examples of well-known enteroviruses are poliovirus (PV), the causative agent of paralytic poliomyelitis, and the human rhinoviruses (HRV), which can cause respiratory tract illnesses and exacerbations of asthma and chronic obstructive pulmonary disease (COPD)1,2. Other clinically important members are echoviruses and coxsackieviruses, which can cause acute clinical manifestations ranging from mild febrile illness to more severe conditions including fulminant sepsis in neonates, meningo-encephalitis, and pancreatitis3. Furthermore, coxsackieviruses have also been associated with chronic diseases, such as chronic myocarditis and type 1 diabetes4. Another significant member of the Enterovirus genus is enterovirus 71 (EV71), which has caused large outbreaks in South East Asia in recent years. Infection with EV71 can result in hand-foot-and-mouth disease, but patients – especially children under 5 years of age – in severe cases develop a potentially fatal central nervous system disease5. Currently, there are no approved antiviral drugs to treat enterovirus infections. Vaccines are only available against PV, yet the WHO campaign for eradication of poliomyelitis is encountering major problems due to the continuous emergence of pathogenic vaccine-derived revertants or recombinants. Development of vaccines against the other enteroviruses is essentially impossible given the large number of (sero)types, exemplified by 150 rhinoviruses, 30 coxsackieviruses, and 30 echoviruses. Thus, there is an urgent need for new tools, such as antiviral drugs, to combat enterovirus infections.
The enterovirus genome encodes four capsid proteins (VP1 through VP4) that facilitate cellular entry and delivery of the viral genome into the cytosol of the host cell, while the seven non-structural proteins (2Apro, 2B, 2C, 3A, 3B, 3Cpro, and 3Dpol) mediate viral RNA replication6. The search for inhibitors of virus replication has resulted in the identification of various compounds that affect the specific stages of the viral life cycle. One of these is enviroxime, a compound already discovered in the late 1970s that inhibits initiation of positive-strand RNA synthesis of PV, HRV, and coxsackievirus B3 (CVB3)7,8,9. Enviroxime-resistant enteroviruses were subsequently isolated in an attempt to identify the target of the compound. Enviroxime-resistant PV, HRV, and CVB3 were all found to carry mutations in 3A, but the resistance mechanism has remained elusive7,8,10. Remarkably, PV mutants that are resistant to GW5074, a kinase inhibitor that inhibits PV RNA replication, were found to contain the same amino acid alteration as mutants resistant to enviroxime11. Intriguingly, we recently identified TTP-8307 as another inhibitor of enterovirus replication and found that TTP-8307-resistant CVB3 contained other mutations in 3A (i.e., V45A and H57Y) that also conferred resistance to enviroxime12.
The enterovirus 3A protein is a small protein that has intrinsic membrane-targeting properties due to a hydrophobic domain in its C-terminus. One of the precursor proteins present during replication is 3AB, of which the presumed function is to provide the primer (3B) for viral RNA synthesis during infection13,14. Additionally, the 3A protein of PV and CVB3 has the ability to perturb the cellular secretory pathway. Under physiological conditions, Arf1, a small GTPase that is activated by the guanine nucleotide exchange factor (GEF) GBF1, is the master regulator of membrane trafficking through the early secretory pathway. Activated Arf1 is responsible for the recruitment of the COP-I complex, a coatomer protein complex that induces membrane curvature and initiates budding of transport vesicles15,16,17. When expressed in isolation, 3A disrupts ER-to-Golgi transport by blocking the formation of COP-I coat complexes on membranes18,19,20,21. The 3A protein most likely exerts this effect by directly interacting with GBF122,23.
The 3A proteins of PV and CVB3 are also responsible for recruiting phosphatidylinositol-4-kinase type III β (PI4KIIIβ), a critical host factor for viral RNA replication, to the sites of RNA replication20. PI4KIIIβ, another downstream effector of Arf1, catalyzes the synthesis of phosphatidylinositol-4-phosphate (PI4P) lipids in Golgi membranes. These PI4P lipids are not only precursors for the classical inositol cycle important for Ca2+ signaling cascades, but have recently also been shown to carry out other functions24,25,26, such as aiding biogenesis and fusion of transport vesicles in the secretory pathway27,28. In enterovirus-infected cells, PI4KIIIβ is actively recruited to the replication sites to create a microenvironment rich in PI4P lipids. The purpose of this environment may be to attract the RNA-dependent RNA polymerase 3Dpol to the replication complex, since 3Dpol specifically binds PI4P over other cellular lipids in vitro20.
In this study, we show that our previously identified enviroxime-resistance mutations in CVB3 3A (i.e., V45A and H57Y) conferred cross-resistance to GW5074 and PIK93, an inhibitor of PI4KIIIβ. Using a genetically encoded PI4P sensor, we furthermore demonstrate that enviroxime and GW5074 strongly reduced the amount of PI4P lipids at the Golgi complex by inhibiting PI4KIIIβ. Next, we set out to investigate how the mutations in 3A provided resistance against these PI4KIIIβ inhibitors. The mutations in 3A did not restore the activity of PI4KIIIβ in the presence of the compounds. Instead, replication of the mutant viruses no longer depended on PI4KIIIβ, nor on any other PI4K isoform, as demonstrated by their efficient replication in cells depleted of these kinases by siRNA-treatment. Finally, we provide evidence that, unlike wild-type (wt) virus, the mutant viruses no longer created a PI4P-rich microenvironment that supports replication. Collectively, our data indicate that CVB3 can bypass an essential host factor for its propagation by a single point mutation in the 3A protein.
Results
Cross-resistance of CVB3 3A mutants to enviroxime, GW5074, and PIK93
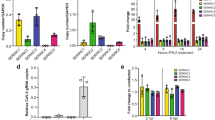
To identify novel inhibitors of CVB3 replication, we screened the Screen-Well Kinase Inhibitor Library, which contains 80 kinase inhibitors. The strongest inhibitor was GW5074, which reduced CVB3 replication by ∼95% (data not shown). GW5074 has previously also been reported to inhibit PV and EV71 replication29. Interestingly, PV mutants resistant against GW5074 contained the same point mutation (i.e., mutation A70T in the hydrophobic domain of 3A) as mutants resistant to enviroxime11. This finding prompted us to test whether our previously identified enviroxime-resistance mutations in CVB3 3A (i.e., mutations V45A and H57Y in the cytosolic domain of 3A) also provided cross-resistance to GW507412. To this end, we infected cells with either wild-type (wt) CVB3 or mutants carrying mutations 3A-V45A or 3A-H57Y for 30 min. After removal of the virus, the different compounds were added to the cells. At 8 h post-infection (p.i.), when the enterovirus life cycle is (nearly) complete, the cells were frozen to release the intracellular virus into the culture medium, after which the virus titer was determined by endpoint titration. Replication of wt virus was inhibited by enviroxime and GW5074 by 3 to 4 logs (Figure 1A). The inhibitory effect of the compounds was not due to cytotoxicity as determined by a cell viability assay (Figure 1C). Both 3A-V45A and 3A-H57Y mutations significantly restored the ability of the virus to replicate in the presence of the compounds (Figure 1A).

CVB3 3A mutants are resistant to enviroxime, GW5074, and PI4KIIIβ inhibitor PIK93. (A, B) BGM cells were infected with CVB3 wt, CVB3 3A-V45A, or CVB3 3A-H57Y (A) or transfected with the corresponding RNA transcripts of full length infectious CVB3 clones (B). Immediately after infection or transfection, enviroxime, GW5074, or PIK93 were added to the cells. After 8 h, cells were lysed by freeze-thawing to release intracellular virus particles and the total virus titer was determined by endpoint titration. (C) BGM or HeLa cells were incubated with the indicated compounds for 8 h using DMSO as a negative control. Subsequently, the cytoxicity of the compounds was determined in a cell viability assay. The percentage was set to 100% for DMSO-treated cells. Bars represent mean of three samples ± SD. Significant differences compared to wt virus are indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001. nd = not detectable.
Recently, we identified PI4KIIIβ as a host factor essential for enterovirus replication and showed that 3A recruits this host factor to the replication sites20. Since GW5074 is a kinase inhibitor and the resistant viruses harbor mutations in the 3A protein, we speculated that the target of GW5074 and enviroxime could be PI4KIIIβ. To study this possibility, we tested whether our 3A mutants were also cross-resistant to PIK93, a well-known and commonly used inhibitor of PI4KIIIβ that can also inhibit PI4KIIIα but with a significantly decreased potency30,31. Figure 1A shows that treatment with PIK93 inhibited replication of wt CVB3 by more than 1 log. Consistent with our speculation, the mutants could replicate significantly better than wt CVB3 in the presence of PIK93. While this work was in progress, a similar observation was made with the enviroxime- and GW5074-resistant PV A70T mutant7,11, which was also found to provide protection against PIK9332.
Resistance of the mutants against the compounds was also observed upon transfection of RNA transcribed from infectious cDNA clones, followed by endpoint titration analysis of the produced virus (Figure 1B). This result demonstrates that the compounds do not affect cell entry or uncoating of the virus. Furthermore, this finding shows that only the mutations in 3A are responsible for the resistance against the compounds.
To demonstrate that our 3A mutations rescued virus replication at the step of viral RNA synthesis, we utilized CVB3-Rluc, a genetically engineered virus that contains Renilla luciferase upstream of the capsid coding region. Infection of cells with CVB3-Rluc virus yields, besides viral proteins, a large amount of Renilla luciferase, which serves as a sensitive measure for viral RNA replication. We treated cells with the compounds after infection with wt or mutant CVB3-Rluc and quantified the intracellular amount of luciferase at various time points p.i. in order to obtain a detailed insight into the RNA replication kinetics (Figure 2). In the absence of compounds, all viruses (i.e., wt and mutants) replicate with similar kinetics in a single cycle (Figure 2A), which is in line with the finding that the mutant viruses induced plaque phenotypes similar to that of wt virus12. Guanidine HCl, a well-known enterovirus inhibitor that blocks viral RNA replication, completely abolished replication of both wt and mutant viruses (Figure 2B). In line with the results of Figure 1, replication of wt CVB3-Rluc was also strongly impaired by the other compounds, whereas the mutant viruses replicated relatively efficiently, albeit with some delay (Figure 2B). Collectively, these results demonstrate that the mutations V45A and H57Y in 3A enable the virus to replicate its RNA in the presence of enviroxime, GW5074, and PIK93.

RNA replication of CVB3 3A mutants in the presence of enviroxime, GW5074, and PIK93. (A, B) HeLa cells were infected with wt or mutant CVB3-Rluc for 30 min. Immediately after infection, Guanidine HCl (GuaHCl), enviroxime, GW5074, or PIK93 were added to the cells (B). Cells were lysed at different time points after infection to determine the intracellular Renilla luciferase activity.
GW5074 and enviroxime decrease PI4P levels on the Golgi complex
The cross-resistance of the 3A mutations to enviroxime, GW5074, and the PI4KIIIβ inhibitor PIK93 suggested that enviroxime and GW5074 also target PI4KIIIβ. Recently, it was demonstrated that GW5074 inhibited PI4KIIIβ directly in an in vitro kinase activity assay32. Here, we tested whether enviroxime is also capable of targeting PI4KIIIβ directly in vitro. Enviroxime exhibited a ∼30-fold preference for PI4KIIIβ over PI4KIIIα and was the most potent inhibitor tested (Figure 3). GW5074 inhibited PI4KIIIβ, but not PI4KIIIα, which is in line with previous results32. This result shows that enviroxime, similar to GW5074, targets PI4KIIIβ directly.

Enviroxime targets PI4KIIIβ directly. Recombinant PI4KIIIβ (A) or PI4KIIIα (B) was incubated with the substrate phosphatidylinositol in the form of Triton micelles and radioactively labeled ATP. After termination of the enzyme reaction, the radioactive ATP incorporated into the micelles is quantified as a measure of PI4K activity. (C) Data were converted to percentage inhibition with respect to positive (wortmannin) and negative (DMSO) controls and also presented as IC50. n.a. = not applicable.
In addition, we evaluated whether enviroxime and GW5074 impair PI4KIIIβ activity in vivo by examining their effects on PI4KIIIβ-catalyzed PI4P synthesis in Golgi membranes. PI4P lipids can be detected in intact cells using a plasmid encoding the PI4P sensor FAPP1-PH-GFP, a GFP-tagged pleckstrin-homology (PH) domain of FAPP1 (four-phosphate-adaptor protein 1). This PH domain contains a PI4P-binding pocket as well as an Arf1-binding site, which together induce localization of this PI4P sensor to the Golgi complex33,34,35,36. In mammalian cells, PI4P lipids present in the early secretory pathway can be produced by three of the four different isoforms of PI4K, namely PI4KIIα (localized to the Golgi), PI4KIIIα (ER), and PI4KIIIβ (Golgi). PI4KIIβ does not localize to the early secretory pathway, but is situated on the plasma membrane and on endosomes24,25. We first investigated which PI4K isoforms are involved in the Golgi-localization of FAPP1-PH-GFP in our cells. To this end, we transfected HeLa cells expressing FAPP1-PH-GFP with siRNAs against PI4KIIα, PI4KIIIα, or PI4KIIIβ. In line with previous results in COS-7 cells33, we observed that PI4KIIIβ is the major determinant of the Golgi-localization of the PI4P sensor (Supplementary information, Figure S1). Thus, FAPP1-PH-GFP can be used to measure the synthesis of PI4P lipids at the Golgi catalyzed by PI4KIIIβ.
Next, we tested the effects of the antiviral compounds on the localization of FAPP1-PH-GFP. One day after transfection with the FAPP1-PH-GFP plasmid, the cells were treated for 1 h with the compounds, after which they were fixed and stained for PI4KIIIβ. In untreated cells, FAPP1-PH-GFP was predominantly localized on the Golgi complex, as shown by the overlap with PI4KIIIβ (Figure 4A). Treatment with either enviroxime, GW5074, or PIK93, however resulted in a severe reduction in the amount of FAPP1-PH-GFP present on the Golgi membranes (Figure 4A). Although analysis of the FAPP1-PH-GFP fluorescence is not the most accurate approach to quantify the effect of the compounds on PI4P levels, we determined the amount of the PI4P sensor present in the Golgi in multiple cells per condition. This quantification revealed that at the used concentrations, enviroxime was consistently the most effective compound (Figure 4B).

Enviroxime and GW5074 decrease PI4P levels at the Golgi complex. (A) HeLa cells were transfected with a plasmid encoding the PI4P sensor FAPP1-PH-GFP. The next day, cells were treated for 1 h with the indicated compounds and stained with an antibody against PI4KIIIβ. (B) Quantification of the percentage of Golgi-localized FAPP1-PH-GFP and PI4KIIIβ, which was determined by dividing the fluorescence intensity of the Golgi-area by that of the area of the whole cell. The resulting percentage was set to 100% in untreated cells. Bars represent the mean of 10 cells from three different experiments ± SD. Significant differences compared to untreated cells are indicated as follows: ***P < 0.001. (C) GBF1 and Arf1 were stained in HeLa cells treated for 1 h with the indicated compounds.
Remarkably, all compounds also induced a significant increase in the intensity of the PI4KIIIβ staining on the Golgi membranes compared to untreated cells (Figure 4A and 4B). This increase in PI4KIIIβ could in theory be the result of activation of upstream factors of PI4KIIIβ that determine its localization. Under physiological conditions, PI4KIIIβ is recruited to the Golgi membranes by the small GTPase Arf137. The activation and consequent membrane association of Arf1 in turn is stimulated by the ArfGEF GBF138. Therefore, we investigated whether the accumulation of PI4KIIIβ on Golgi membranes was due to effects of the compounds on either GBF1 or Arf1 using an immunofluorescence assay. The localization as well as the amount of GBF1 and Arf1 on Golgi membranes was not altered after a 1-h treatment with the compounds (Figure 4C), indicating that it is unlikely that the compounds affect these upstream regulators of PI4KIIIβ. Hence, the increase in PI4KIIIβ on the Golgi complex is most likely caused by an unknown compensatory mechanism, possibly involving phosphorylation of PI4KIIIβ, of the cell39. In conclusion, these results collectively suggest that both enviroxime and GW5074 diminish PI4P levels on the Golgi through inhibition of PI4KIIIβ activity.
Rescue of CVB3 replication in the presence of enviroxime by expression of a PIK93-resistant PI4KIIIβ
GW5074 targets a broad and not yet fully defined range of cellular kinases40, which may also be the case for enviroxime. Therefore, targets other than PI4KIIIβ could contribute to the inhibitory effect of both compounds on enterovirus replication. To test whether PI4KIIIβ is the only (major) target of the compounds responsible for their antiviral activity, we conducted a “replication rescue assay” using a PI4KIIIβ mutant resistant to the PI4KIIIβ inhibitors wortmannin and PIK9330. This PI4KIIIβ mutant contains a single mutation (Y583M) that weakens the binding of wortmannin and PIK93 in the ATP-binding pocket without affecting the catalytic activity of the kinase. Cells were transfected with either PI4KIIIβ-wt, PI4KIIIβ-Y583M, or as negative controls the kinase-dead PI4KIIIβ-D656A or EGFP. Two days post-transfection, the cells were infected with wt CVB3 or CVB3-Rluc in the presence of the compounds. At 8 h p.i., cells were lysed to determine either the total virus titer or the intracellular amount of luciferase. Similar expression levels of all PI4KIIIβ proteins were confirmed in parallel in an immunofluorescence assay (data not shown). Figure 5A shows that transfection of PI4KIIIβ-wt rescued the replication of CVB3-Rluc in the presence of the compounds only to a marginal extent. As expected, EGFP and the kinase-dead PI4KIIIβ-D656A could not counter the effects of the compounds on CVB3 replication. The presence of PI4KIIIβ-Y583M however markedly restored CVB3 replication in the presence of either PIK93 or enviroxime nearly to untreated levels.

Mutant PI4KIIIβ rescues the effect of enviroxime on CVB3 replication. (A) Replication rescue experiment. BGM cells were transfected with HA-tagged PI4KIIIβ-wt, PI4KIIIβ-Y583M, or as negative controls the kinase-dead PI4KIIIβ-D656A or EGFP. Two days post-transfection, cells were infected with CVB3 or CVB3-Rluc in the presence of the indicated compounds. After lysing the cells at 8 h p.i., the total virus titer or the amount of luciferase activity was determined in the samples. (B, C) HeLa cells were co-transfected with plasmids encoding FAPP1-PH-GFP and either HA-tagged PI4KIIIβ-wt (B) or PI4KIIIβ-Y583M (C). The next day, cells were treated for 1 h with the indicated compounds. Subsequently, cells were fixed and the overexpressed PI4KIIIβ was stained using an antibody against HA. The percentage of Golgi-localized FAPP1-PH-GFP was determined by dividing the fluorescence intensity of the Golgi-area by that of the area of the whole cell. The resulting percentage was set to 100% in untreated cells. Bars represent the mean of 10 cells from three different experiments ± SD. Significant differences compared to untreated cells are indicated as follows: **P < 0.01, ***P < 0.001.
The effects of GW5074 on the other hand were not countered by PI4KIIIβ-Y583M, for which the explanation could be that the Y583M mutation does not provide resistance to GW5074. To test this possibility, we co-transfected the plasmids encoding FAPP1-PH-GFP with either PI4KIIIβ-wt or PI4KIIIβ-Y583M and evaluated the effect of the compounds on the presence of the PI4P sensor on the Golgi. In cells expressing PI4KIIIβ-wt, all compounds strongly reduced the presence of the PI4P sensor on Golgi membranes (Figure 5B). Expression of PI4KIIIβ-Y583M however only restored the Golgi-localization of the PI4P sensor for enviroxime and PIK93 (Figure 5C). In line with the observation that PI4KIIIβ-Y583M could not rescue CVB3 replication in the presence of GW5074, PI4KIIIβ-Y583M was not resistant to GW5074 as shown by the absence of FAPP1-PH-GFP on the Golgi (Figure 5C). Taken together, these data suggest that PI4KIIIβ is the major target of enviroxime and PIK93 that is responsible for the inhibition of CVB3 replication.
Mutations in 3A do not rescue the activity of PI4KIIIβ in the presence of enviroxime, GW5074, and PIK93
Next, we investigated how the mutations in 3A confer resistance to enviroxime, GW5074, and PIK93. One could speculate that mutations in 3A could somehow rescue the activity of PI4KIIIβ in the presence of the compounds. Another possible explanation for the resistant phenotype is that the mutants replicate independently of PI4KIIIβ. To test the first scenario, we studied the effect of the compounds on PI4P levels in cells expressing either wt or mutant 3A. To this end, we co-transfected cells with plasmids encoding FAPP1-PH-GFP together with wt or mutant 3A-myc, and one day later treated them for 1 h with the compounds. In untreated cells, wt 3A was visible in a punctate pattern (Figure 6A), corresponding to a localization to dispersed Golgi membranes, as previously described20,23. 3A showed a great overlap with PI4P, as detected by FAPP1-PH-GFP. In cells treated with enviroxime, GW5074, or PIK93, little if any FAPP1-PH-GFP was present on 3A-containing membranes (Figure 6A), which indicates that wt 3A is unable to prevent or restore the compound-induced inhibition of PI4KIIIβ activity. In cells expressing 3A-H57Y-myc (Figure 6B) or 3A-V45A-myc (data not shown), treatment with the compounds also resulted in a nearly complete absence of FAPP1-PH-GFP on 3A-containing membranes. The reduction in PI4P levels is not due to an absence of PI4KIIIβ, which was recruited equally well by wt 3A, 3A-V45A (not shown), and 3A-H57Y in the presence of the compounds (Supplementary information, Figure S2). These results suggest that it is unlikely that the resistance mutations in 3A prevent or counter the effect of the compounds on PI4KIIIβ activity.

3A-H57Y does not counter the decrease in PI4P levels induced by enviroxime, GW5074, and PIK93. HeLa cells were co-transfected with plasmids encoding FAPP1-PH-GFP and myc-tagged wt 3A (A) or 3A-H57Y (B). One day after transfection, cells were treated for 1 h with compounds. Subsequently, 3A and 3A-H57Y were stained with an anti-myc antibody. The FAPP1-PH-GFP to 3A fluorescence was quantified in 10 cells per condition and expressed as a ratio. Bars represent the mean ± SD. Significant differences compared to untreated cells are indicated as follows: ***P < 0.001.
CVB3 3A mutants replicate efficiently in cells depleted of PI4KIIIβ
Next, we investigated whether the mutants still require PI4KIIIβ for replication. To this end, we first investigated whether the mutant viruses also recruit PI4KIIIβ to the sites of RNA replication as wt virus does. Cells were infected with either wt or mutant viruses carrying mutations V45A or H57Y, fixed at 5 h p.i., and stained for PI4KIIIβ and dsRNA, a generally accepted marker of viral replication sites. For all viruses, an intense staining of PI4KIIIβ was observed at the replication sites (Figure 7A, CVB3 3A-V45A not shown), which demonstrates that all viruses induced an active recruitment of PI4KIIIβ to the replication sites. PI4KIIIβ was also present on the replication sites of mutant viruses induced in the presence of the compounds (data not shown), which is consistent with the observation that the compounds also did not alter the PI4KIIIβ recruitment in cells transfected with wt or mutant 3A (Supplementary information, Figure S2).

Replication of CVB3 3A mutants is largely independent of PI4KIIIβ. (A) HeLa cells were infected with wt CVB3 or mutant CVB3 3A-H57Y. At 5 h p.i., cells were fixed and stained with antibodies against PI4KIIIβ and dsRNA as a marker for the sites of RNA replication. (B, C) HeLa cells were transfected with siRNAs against PI4KIIIβ (B), or PI4KIIα, PI4KIIβ, and PI4KIIIα (C) and scrambled siRNA as a negative control. Two days later, siRNA-treated cells were infected with wt virus, 3A-V45A virus, or 3A-H57Y virus. Virus titers were determined at 8 h p.i. Bars represent mean of three samples ± SD, *P < 0.05. (D) In parallel, efficiency of the knock-down was determined by western blot analysis.
The presence of PI4KIIIβ on the replication sites induced by the mutant viruses does not necessarily prove that this host factor is essential for their replication. We next investigated whether PI4KIIIβ is critical for replication of the mutant viruses by testing the ability of the mutants to replicate in cells depleted of PI4KIIIβ by siRNA-treatment. Cell viability assays showed that the knock-down was not toxic for the cells (Supplementary information, Figure S3). Western blot analysis of control- and siRNA-treated cells revealed a knock-down efficiency of ∼90-95% (Figure 7D). Consistently, IFA analysis showed that 5-10% of the cells were not transfected with siRNAs against PI4KIIIβ and, hence, showed normal expression levels of this protein (data not shown). These untransfected cells can still support CVB3 replication and thereby a maximal decrease in virus titer of 1 to 1.5 log can be achieved. In agreement with this transfection efficiency, we observed that replication of wt CVB3 was reduced by ∼1.2 log in PI4KIIIβ-depleted cells (Figure 7B). Surprisingly, replication of both mutants was not affected upon knock-down of PI4KIIIβ, which demonstrates that replication of the mutants is largely independent of PI4KIIIβ.
Mutations in 3A render CVB3 replication independent of high levels of PI4P lipids
A possible strategy of cells and viruses to overcome inhibition of crucial factors is to switch to other isoforms that are functionally redundant. Hence, we investigated whether the mutant viruses utilized PI4K isoforms other than PI4KIIIβ for the production of PI4P. To this end, we tested whether replication of wt or mutant CVB3 was affected by depletion of PI4KIIα, PI4KIIβ, or PI4KIIIα by siRNA-treatment. Knock-down of these proteins was efficient and not toxic for the cells (Figure 7D, Supplementary information, Figure S3 and S4). Figure 7C shows that replication of neither wt nor mutant viruses was affected by knock-down of PI4KIIα, PI4KIIβ, or PI4KIIIα. This result furthermore suggests that the mutant viruses do not rely on high levels of PI4P for replication, since they do not require any isoform of PI4K.
Next, we investigated whether the replication sites induced by the CVB3 mutants in the presence of the compounds indeed were devoid of PI4P lipids. Initially, we opted for transfection of cells with the plasmid encoding FAPP1-PH-GFP followed by virus infection and compound-treatment the next day. However, this method resulted in very low numbers of cells that were both transfected, as well as efficiently infected. Therefore, we switched to an immunofluorescence assay for intracellular staining of PI4P lipids using an antibody that specifically recognizes PI4P lipids. Cells were infected with either wt or mutant viruses in the presence or absence of the compounds, fixed at 5 h p.i., and stained for 3A and PI4P. We did not detect expression of 3A in Guanidine HCl-treated cells (data not shown), indicating that virus replication is required to generate detectable amounts of 3A. Upon replication, both CVB3 wt and the 3A-V45A and -H57Y mutant viruses induced a dramatic increase in PI4P levels in the absence of the compounds (Figure 8, CVB3 3A-V45A not shown). No infected cells were detected for wt CVB3 in the presence of the compounds, since they inhibit replication (data not shown). Consistent with the results of Figures 1 and 2, replication of the mutant viruses was slightly delayed by the compounds, as shown here by the reduced amount of infected cells. Accordingly, the pattern of 3A was in some cells more typical for the early stage of replication, when the distribution of 3A is still restricted to the Golgi area20. Remarkably, the PI4P lipids appeared absent in cells infected with the mutant viruses in the presence of enviroxime and GW5074 (Figure 8). Importantly, also no high levels of PI4P lipids were detected in these cells at later time points, when cells started to round up and display cytopathic effects (data not shown). In cells treated with PIK93 however, some PI4P lipids could still be detected, which is consistent with our observation that PIK93 at the used concentration was the weakest inhibitor of PI4KIIIβ. These observations imply that the mutant viruses can establish replication without the participation of high PI4P levels. Collectively, these data suggest that the mutations 3A-V45A and 3A-H57Y render virus replication largely independent of host factor PI4KIIIβ as well as PI4P lipids.

CVB3 3A mutants do not require a high level of PI4P lipids. HeLa cells were infected with CVB3 wt or CVB3 3A-H57Y, after which the medium was replaced with (compound-containing) medium. At 5 h p.i., cells were fixed and stained with antibodies against 3A and PI4P. The PI4P to 3A fluorescence was quantified in 10 cells per condition and expressed as a ratio. Bars represent the mean ± SD. Significant differences compared to untreated cells are indicated as follows: ***P < 0.001.
Discussion
The emergence of drug-resistant viruses is a common response to drugs targeting viral proteins due to the genetic plasticity of RNA viruses. To circumvent this problem, there is a great interest in the development of antiviral drugs that target host factors, since these factors are unlikely to mutate in response to therapy. However, this “host-oriented” approach is also based on the assumption that viruses are not capable of mutating in such a way that they can overcome drug-induced inhibition of a critical host factor. Here, we present a unique example of how viruses by acquiring a single point mutation can bypass a critical host factor and replicate efficiently when this factor is inhibited or depleted by siRNA-treatment.
While the development of viral drug-resistance poses serious problems in a clinical context, it is a useful tool in studies on the mechanism of action of antiviral compounds. The CVB3 mutants studied here, 3A-V45A and 3A-H57Y, exhibited resistance to enviroxime and GW5074 as well as the PI4KIIIβ inhibitor PIK93, which suggested that these compounds also target PI4KIIIβ. Consistent with this idea, both enviroxime and GW5074 inhibited PI4KIIIβ directly in an in vitro kinase activity assay. Furthermore, both compounds caused a decrease in localization of the PI4P sensor FAPP1-PH-GFP at the Golgi complex, reflecting a reduction in PI4P levels. Since PI4KIIIβ was the major determinant of the Golgi-localization of the PI4P sensor in our cells, this result indicated that enviroxime and GW5074 also inhibited PI4KIIIβ activity in intact cells. Furthermore, CVB3 replication in the presence of enviroxime was restored nearly to levels in untreated cells by the expression of a PI4KIIIβ mutant. This PI4KIIIβ mutant carries a mutation in its ATP-binding pocket, Y853M, that does not harm the catalytic activity of the kinase. The rescue of the effects of enviroxime on CVB3 replication by this PI4KIIIβ mutant has three implications. First, the compound only inhibits the enzymatic activity of PI4KIIIβ required for CVB3 replication and not a non-enzymatic function. Second, PI4KIIIβ and not a yet unidentified target of enviroxime is responsible for the inhibition of CVB3 replication. Third, enviroxime, similar to PIK93, binds in the ATP-binding pocket of PI4KIIIβ.
Our observation that GW5074 inhibits PI4KIIIβ activity in vitro is in line with the results of a recent study, which was published while our work was in progress32. Here, we also show that GW5074 targets PI4KIIIβ in intact cells by use of a PI4P-sensor. Besides PI4KIIIβ, GW5074 inhibits a variety of additional kinases other than its best-known target c-Raf, such as the kinases Pim-1, Pim-2, Pim-3, HIPK2, RIP2, GAK, and MST240, and therefore represents a typical example of a promiscuous kinase inhibitor. Importantly, previous studies had already excluded a role of these other GW5074-targeted kinases in enterovirus replication29. The effects of GW5074 on CVB3 replication could not be countered by PI4KIIIβ-Y583M, which indicates that binding of this compound to PI4KIIIβ does not involve Y583 in the ATP-binding pocket. The inhibition of kinases by GW5074 can occur in a competitive manner with GW5074 acting as an ATP/GTP analog41,42, or alternatively in a non-competitive manner, as exemplified by the prevention of hexamer formation of glutamate dehydrogenase43,44. The mechanism by which GW5074 inhibits PI4KIIIβ remains to be elucidated in future studies.
After demonstrating that enviroxime and GW5074 inhibit PI4KIIIβ activity in vitro and in intact cells, we next investigated how the 3A mutations provide compound resistance. One possible explanation for the drug-resistant phenotype is that the 3A-V45A and 3A-H57Y mutations rescued the activity of PI4KIIIβ in compound-treated cells. However, we excluded this possibility by showing that in cells expressing either wt or mutant 3A, the compounds still induced a strong reduction in the localization of FAPP1-PH-GFP on 3A-containing membranes. Thereafter, we considered the scenario that the mutations rendered viral RNA replication independent of PI4KIIIβ. Indeed, replication of the mutant viruses was no longer sensitive to knock-down of PI4KIIIβ. Additionally, we observed efficient replication upon knock-down of the isoforms PI4KIIα, PI4KIIβ, and PI4KIIIα, indicating that the mutants did not rely on any isoform of PI4K. This finding raised the question of whether the mutant viruses still needed PI4P for replication. An immunofluorescence assay in compound-treated infected cells with an antibody against PI4P revealed that the mutant viruses were indeed able to replicate in cells lacking detectable PI4P levels. However, we cannot exclude the possibility that there is still a tiny, undetectable amount of PI4P present in these cells on which the mutant virus might depend.
The purpose of PI4P lipids in enterovirus replication is currently unknown. Under physiological conditions, PI4P lipids were for a long time merely perceived as precursors for the production of PIP2 and IP3 in the classical inositol cycle important for intracellular Ca2+ signaling24. However, PIP2 levels at the membranes supporting replication were unaltered and even decreased along the course of CVB3 infection20. Recently, PI4P lipids were demonstrated to have additional important functions in the early secretory pathway through their ability to serve as docking sites for various proteins24,26. Consistent with this ability of PI4P, the RNA-dependent RNA polymerase 3Dpol of CVB3 was shown to bind preferentially to PI4P lipids over other cellular lipids in vitro20. This finding suggests that the purpose of PI4P in enterovirus replication might be to recruit 3Dpol to the sites of viral RNA replication and/or to stimulate the activity of 3Dpol, yet evidence that this occurs in enterovirus-infected cells is lacking.
Another speculative scenario for the role of PI4P lipids during enterovirus infection is that they recruit other cellular factors important for replication. PI4P lipids are bound by proteins that contain a pleckstrin homology (PH) domain. Several Golgi-proteins that contain a PI4P-binding PH domain are involved in lipid transport, such as FAPP1, oxysterol-binding protein (OSBP) and ceramide transport protein (CERT)33,36,45. The primary function of these proteins is to bind ceramide, cholesterol, and/or oxysterols (i.e., oxygenated derivatives of cholesterol) to mediate non-vesicular transport of these lipids to other compartments in the cell. Therefore, recruitment of these lipid-binding proteins to the enterovirus replication sites might lead to accumulation of other specific lipids other than PI4P required for viral RNA replication and/or membrane reorganization.
Recently, our view of the roles of lipids in viral infection has been greatly expanded by the recognition that positive-strand RNA viruses actively alter cellular lipid signaling, synthesis, and metabolism for formation and function of their replication sites. Similar to enteroviruses, the flavivirus Hepatitis C virus (HCV) requires PI4P lipids for replication, which are generated via the recruitment of PI4KIIIα46,47,48,49,50,51,52,53. West Nile virus attracts the cholesterol-synthesizing enzyme 3-HMGA-CoA reductase to replication sites54,55, while dengue virus recruits fatty acid synthase and stimulates its activity56,57. The recruitment of lipid biosynthesis machinery can lead to membrane expansion and alteration of the fluidity and curvature of the membrane by for instance accumulation of cholesterol or desaturation of lipid fatty acid tails. The consequences of lipid synthesis and modification may be important for the membrane reorganization that positive-strand RNA viruses induce to create their own membranous environment with a highly specialized lipid composition suited for viral RNA replication58. Given the accumulating evidence that pre-existing cellular membranes have to be expanded and/or modified to accommodate the replication complexes of positive-strand RNA viruses, and that modulation of cellular lipid metabolism appears to be a universal feature of this group of viruses, it is remarkable that we have identified a mutant virus that can efficiently replicate in the absence of an essential lipid. To our knowledge, this is the first example of a positive-strand RNA virus that can replicate in the absence of high levels of a crucial lipid in the environment of their replication sites, on which the wt virus ultimately depends. Currently, we are investigating whether the absence of PI4P affects the three-dimensional architecture of the enterovirus membranous replication sites.
Since the role of PI4P lipids in enterovirus replication remains elusive, it is difficult to explain how our CVB3 mutants can replicate in the absence of high levels of PI4P lipids, albeit somewhat less efficiently than when PI4P lipids are present. Replication of the wt virus is completely abolished when PI4KIIIβ is inhibited by enviroxime and is also strongly impaired upon depletion of this host factor by siRNA-treatment. Since replication of the mutant virus was largely unaffected upon inhibition or depletion of PI4KIIIβ, the mutant virus may be able to use an alternative pathway without PI4KIIIβ and PI4P to establish replication. Although the single point mutations allow CVB3 to replicate in cells lacking PI4KIIIβ and PI4P lipids, we speculate that the PI4KIIIβ-dependent pathway offers an evolutionary advantage over the alternative pathway. All CVB3 strains isolated to date contain V45 and H57 in their 3A sequence, suggesting a selective pressure on the usage of PI4KIIIβ and PI4P lipids. Moreover, we found that replication of all tested enteroviruses, including various serotypes belonging to different human enterovirus and rhinovirus species, is strongly inhibited by enviroxime12, which further illustrates the functional importance of PI4KIIIβ and PI4P lipids in their replication cycle. In future studies, it will be of interest to dissect the alternative pathway used by the 3A mutant viruses. Intriguingly, the CVB3 resistance mutations 3A-V45A and 3A-H57Y are located in the cytosolic domain, whereas mutation 3A-A70T that rendered PV resistant to enviroxime, GW5074, and PIK93 is located in the hydrophobic, membrane-anchored domain. How these mutations enable the virus to replicate in the absence of active PI4KIIIβ remains to be established.
The CVB3 3A mutants are the first example of picornaviruses able to replicate without the participation of a crucial host factor. Previously, PV mutants were generated that were resistant to Brefeldin A59, a well-known inhibitor of enterovirus replication that perturbs secretory pathway transport by targeting GBF1. These PV mutants contained the mutations V80I in 2C and A27V in 3A. PV carrying the single mutations exhibited intermediate resistance to BFA, while the double-mutant was fully resistant. Despite the insensitivity to Brefeldin A, however, the PV double-mutant could not replicate in cells depleted of the crucial factor GBF1 by siRNA-treatment60.
Virus replication largely independent of a host factor has been described before for another positive-strand RNA virus. An HCV mutant resistant against Alisporivir and cyclosporine A was previously isolated that contained a point mutation in NS5A. Alisporivir and cyclosporine A exert their antiviral activity by binding to cyclophilins, which are critical host factors for HCV. Cyclophilins affect the folding of proteins through their peptidyl-prolyl cis-trans isomerase activity, which catalyzes the cis-trans isomerisation of the prolyl peptide bond preceding proline residues. Due to the resistance mutation, HCV NS5A adopted a conformation that was no longer dependent on the cis-trans isomerisation activity of the cyclophilins for its function61. These examples of mutant HCV and CVB3 demonstrate that viruses can acquire resistance even to compounds that target crucial host factors. Furthermore, they disclose the ability of viruses to use an alternative pathway to replicate efficiently in a suboptimal environment under selective pressure imposed by antiviral compounds, suggesting that a combination of compounds targeting different host factors may be required for successful host-oriented antiviral therapy.
Materials and Methods
Cells and reagents
Buffalo green monkey (BGM) kidney cells and HeLa (R19) cells were grown at 37 °C, 5% CO2 in minimal essential medium (MEM) (Gibco) supplemented with 10% fetal bovine serum, penicillin, and streptomycin. Enviroxime was synthesized as described previously9. Guanidine hydrochloride and GW5074 were purchased from Sigma Aldrich. PIK93 was provided by Dr K Shokat (University of California San Francisco, San Francisco, California, USA). All compounds were dissolved in DMSO. GW5074 was used at a concentration of 30 μM, enviroxime at 0.5 μg/ml (1.4 μM), and PIK93 at 1 μM.
Antibodies
As primary antibodies we used rabbit polyclonal anti-PI4-KinaseIIIβ (Upstate), mouse monoclonal anti-GBF1 (BD Biosciences), rabbit polyclonal anti-Arf1 (kind gift from dr. F. Wieland, Biochemie-Zentrum Heidelberg, Germany), mouse monoclonal anti-c-Myc (Sigma Aldrich), mouse monoclonal anti-dsRNA (J2, English & Scientific Consulting), mouse anti-PI4P IgM (Echelon Biosciences), mouse monoclonal anti-HA (Covance), mouse monoclonal anti-β-actin (Sigma), and rabbit polyclonal anti-CVB3 3A described previously23. The secondary antibodies for immunofluorescence included Alexa Fluor 488- or 594-conjugated goat-anti-rabbit IgG, Alexa Fluor 488- or 594-conjugated goat-anti-mouse IgG, or Alexa Fluor 594-conjugated goat-anti-mouse IgM (Molecular Probes). The secondary antibodies for Western Blot analysis included IRDye anti-mouse or anti-rabbit antibodies (Li-Cor Biosciences).
Viruses
CVB3 and CVB3-Rluc, which contains the Renilla luciferase gene upstream of the capsid coding region, were obtained by transfection of RNA transcripts derived from the full length infectious clones p53CB3/T7 and pRLuc-53CB3/T7 in BGM cells as described before21,62. CVB3(-Rluc) carrying the V45A or H57Y mutation in the 3A protein were obtained in a similar manner using plasmids p53CB3/T7-3A-V45A or -H57Y described elsewhere12 or pRluc-53CB3/T7-3A-V45A or -H57Y. Virus titers were determined by endpoint titration according to the method of Reed and Muench and expressed as 50% cell culture infective doses (CCID50)63.
Plasmids
The plasmid p3A-myc and the p53CB3/T7 3A mutant full length infectious clones have been described previously12,21. To construct the plasmids p3A-V45A-myc and p3A-H57Y-myc, the 3A gene containing the V45A or H57Y mutation was amplified using the mutant full-length clones as a template. The forward primer introduced a SalI restriction site and an ATG codon (5′-GGG GGG TCG ACC ATG GGA CCA CCA GTA TAC AGA-3′), and the reverse primer introduced a BamHI site (5′-GGG GGG GGA TCC TTG AAA ACC CGC AAA GAG-3′). The PCR product was cloned into p3A-myc using restriction enzymes SalI and BamHI. To construct pRLuc-53CB3/T7 containing the mutations 3A-V45A or 3A-H57Y for the production of mutant CVB3-Rluc, p53CB3/T7 containing the mutations was digested with XbaI and BstEII. The insert encoding the region of 3A with the mutations was ligated into pRLuc-53CB3/T7. The presence of the mutations in all constructs and produced virus was confirmed by sequence analysis. The plasmids encoding FAPP1-PH-GFP, PI4KIIIβ-HA, PI4KIIIβ-Y583M-HA, and kinase-dead PI4KIIIβ-D656A were kindly provided by Dr T Balla (NICHD, National Institutes of Health, Bethesda, Maryland, USA)33,64. The plasmid encoding PI4KIIβ-myc to verify the knock-down efficiency by siRNA-treatment was generously provided by Dr J Albanesi (UT Southwestern Medical Center, Dallas, Texas, USA)65.
Kinase library screen
The Screen-Well Kinase Inhibitor Library containing 80 kinase inhibitors was purchased from Biomol. One hour prior to infection, the compounds were added to HeLa cells – seeded in 96-well plates – to a final concentration of 10 μM. DMSO was used as a negative control. Subsequently, cells were infected with CVB3-Rluc virus at low MOI. After lysing the cells at 8 h post-infection (p.i.), the intracellular Renilla luciferase activity was determined with the Renilla Luciferase Assay System (Promega) according to the manufacturer's instructions.
Virus infections
Subconfluent layers of cells in 24-well plates were infected with CVB3(-Rluc) at an MOI of 0.1-1. After 30 min virus adsorption, cells were washed and fresh (compound-containing) medium was added to the cells. At 8 h p.i., cells were subjected to three cycles of freeze-thawing after which virus titers were determined by endpoint titration. Alternatively, cells were lysed to determine the intracellular Renilla luciferase activity with the Renilla Luciferase Assay System (Promega).
RNA transfection
The full length infectious clones of wt CVB3 p53CB3/T7 and the 3A mutants (V45A, and H57Y) were linearized with SalI, purified and transcribed in vitro by T7 RNA polymerase. The RNA was transfected into BGM cells in a 24-well plate using the DEAE-dextran method as described previously66. After transfection, cells were treated with compounds when indicated, and incubated at 37 °C. At 8 h post-transfection, the cells were lysed by three rounds of freeze-thawing, after which the samples were subjected to titration analysis.
Immunofluorescence assay
HeLa cells were grown to subconfluency on coverslips in 24-well plates. When indicated, cells were transfected with 300 ng DNA per construct using FuGENE (Roche) according to the manufacturer's protocol. After 16 h, cells were treated with compounds for on1 h at 37 °C. Subsequently cells were fixed with 4% paraformaldehyde and permeabilized with PBS containing 0.1% Triton X-100. In case of the PI4P antibody, 0.2% saponin was used to stain only intracellular PI4P. Subsequently, cells were stained with primary and fluorescently-labeled secondary antibodies. Nuclei were stained with Hoechst. Cells were analyzed with a wide-field Leica BMR microscope at 400× magnification. Hoechst, GFP/Alexa Fluor 488, or Alexa Fluor 594 were excited through bandpass filters of 360/20 nm, 470/20 nm, or 540/20 nm, respectively. Longpass filters of 425 nm (Hoechst), 515 nm (GFP/Alexa Fluor 488), or 590 nm (Alexa Fluor 594) were used for the fluorescence emission. Fluorescent images of typical examples of observations that we consistently obtained in three or more experiments are presented in the figures. Quantification was performed per condition on 10 cells from different experiments using ImageJ software. For the quantification of the amount of Golgi-localized FAPP1-PH-GFP and PI4KIIIβ, the fluorescence intensity of the Golgi-area was divided by that of the area of the whole cell. The resulting percentage was set to 100% in untreated cells. For the quantification of FAPP1-PH-GFP localized to 3A-containing membranes, we determined the fluorescence intensities of FAPP1-PH-GFP and 3A(-H57Y)-myc in the myc-stained areas and expressed this as a ratio. To quantify the PI4P levels in infected and compound-treated cells, we determined the ratio between the fluorescence intensities of PI4P and 3A in the whole cell.
PI4K in vitro activity assay
A radioactive filter plate assay was used to measure the in vitro activity of the compounds on PI4KIIIα and PI4KIIIβ. Fivefold serial dilutions of compounds were added to the wells. Recombinant PI4KIIIβ (SignalChem) and the substrate phosphatidylinositol (PI): phosphatidylserine (PS) (2 mM PI:8 mM PS) in 0.3% Triton X-100 were diluted in 20 mM Tris pH 7.5, 5 mM MgCl2, 5 mM EGTA, 2 mM DTT, 0.04% Triton X-100 to a final concentration of 400 ng/ml and 75 μM, respectively. For PI4KIIIα (Millipore), the enzyme and the substrate were diluted in 20 mM MOPS pH 7.0, 50 mM NaCl, 2 mM MgCl2, 1 mM DTT, 0.04% Triton X-100 to a final concentration of 300 ng/ml and 15 μM, respectively. The reaction was started by addition of 1-5 μM ATP and 0.25 μCi 33P]- γ-ATP, and the mixture was incubated at 30 °C for 75-90 min. After termination of the reaction with phosphoric acid, the samples were transferred to filter plates and incorporated radioactivity was measured by TopCount NXT™ Microplate Scintillation (PerkinElmer). Data were converted to percent inhibition with respect to positive (wortmannin) and negative (DMSO) controls.
Replication rescue assay
BGM cells were seeded in 96-well plates and transfected with pEGFP (negative control), pPI4KIIIβ-HA, pPI4KIIIβ-Y583M-HA, or kinase-dead PI4KIIIβ-D656A (negative control) on the next day. Two days post-transfection, the cells were infected with CVB3-Rluc at an MOI of 0.1 for 30 min. After virus removal, fresh (compound-containing) medium was added to the cells. At 8 h p.i., cells were lysed and the intracellular Renilla luciferase activity was determined with the Renilla Luciferase Assay System (Promega). Similar expression levels of the proteins were confirmed in parallel in an immunofluorescence assay using an antibody directed against HA.
siRNA-treatment
HeLa cells were reversely transfected with 1 pmol of siRNA per well of a 96-well plate using RNAiMAX (Invitrogen) according to the manufacturer's protocol. Scrambled siRNA was obtained from QIAGEN, while the siRNAs targeting PI4KIIα, PI4KIIIα or PI4KIIIβ were purchased from Ambion. At 48 h after siRNA-treatment, cells were infected with either wt CVB3, CVB3 3A-V45A, or CVB3 3A-H57Y at an MOI of 0.1. At 8 h p.i., cells were freeze-thawed three times to determine total virus titers by end-point titration. To verify efficient knock-down of the targets by the siRNA treatment, cells of 6 wells were harvested and pooled using TEN buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 0.1 M NaCl). Following cell lysis, proteins were analyzed by SDS-PAGE (10% polyacrylamide). The proteins were blotted on nitrocellulose membranes (Bio-Rad) and stained with primary and secondary antibodies. Imaging was done with the Odyssey System. To verify the knock-down efficiency of PI4KIIβ, cells were transfected 24 h after siRNA-treatment with a plasmid encoding PI4KIIβ-myc. At 48 h after siRNA-treatment, cells were fixed and stained with an anti-myc antibody.
Cell viability assay
One day after seeding cells in a 96-well plate, the compounds were added to the cells and incubated for 8 h. Alternatively, cells were transfected with siRNAs and incubated for 48 h. Subsequently, the medium was replaced with CellTiter 96 AQueous One Solution Reagent (Promega) and optical densities were measured at 490 nm. The obtained densities were corrected for background absorbance from wells that lacked cells. The resulting percentage was set to 100% for DMSO-treated cells or for cells that were not transfected with siRNAs.
References
Gern JE . The ABCs of rhinoviruses, wheezing, and asthma. J Virol 2010; 84:7418–7426.
Mallia P, Contoli M, Caramori G, Pandit A, Johnston SL, Papi A . Exacerbations of asthma and chronic obstructive pulmonary disease (COPD): focus on virus induced exacerbations. Curr Pharm Des 2007; 13:73–97.
Tebruegge M, Curtis N . Enterovirus infections in neonates. Semin Fetal Neonatal Med 2009; 14:222–227.
Whitton JL, Cornell CT, Feuer R . Host and virus determinants of picornavirus pathogenesis and tropism. Nat Rev Microbiol 2005; 3:765–776.
Bible JM, Pantelidis P, Chan PK, Tong CY . Genetic evolution of enterovirus 71: epidemiological and pathological implications. Rev Med Virol 2007; 17:371–379.
Bedard KM, Semler BL . Regulation of picornavirus gene expression. Microbes Infect 2004; 6:702–713.
Heinz BA, Vance LM . The antiviral compound enviroxime targets the 3A coding region of rhinovirus and poliovirus. J Virol 1995; 69:4189–4197.
Heinz BA and Vance LM . Sequence determinants of 3A-mediated resistance to enviroxime in rhinoviruses and enteroviruses. J Virol 1996; 70:4854–4857.
Wikel JH, Paget CJ, DeLong DC, et al. Synthesis of syn and anti isomers of 6-[[(hydroxyimino)phenyl]methyl]-1-[(1-methylethyl)sulfonyl]-1H-benzimidazol-2-amine. Inhibitors of rhinovirus multiplication. J Med Chem 1980; 23:368–372.
Brown-Augsburger P, Vance LM, Malcolm SK, Hsiung H, Smith DP, Heinz BA . Evidence that enviroxime targets multiple components of the rhinovirus 14 replication complex. Arch Virol 1999; 144:1569–1585.
Arita M, Wakita T, Shimizu H . Cellular kinase inhibitors that suppress enterovirus replication have a conserved target in viral protein 3A similar to that of enviroxime. J Gen Virol 2009; 90:1869–1879.
De Palma AM, Thibaut HJ, van der Linden L, et al. Mutations in the nonstructural protein 3A confer resistance to the novel enterovirus replication inhibitor TTP-8307. Antimicrob Agents Chemother 2009; 53:1850–1857.
Liu Y, Franco D, Paul AV, Wimmer E . Tyrosine 3 of poliovirus terminal peptide VPg(3B) has an essential function in RNA replication in the context of its precursor protein, 3AB. J Virol 2007; 81:5669–5684.
Towner JS, Mazanet MM, Semler BL . Rescue of defective poliovirus RNA replication by 3AB-containing precursor polyproteins. J Virol 1998; 72:7191–7200.
Bonifacino JS, Glick BS . The mechanisms of vesicle budding and fusion. Cell 2004; 116:153–166.
Lee MC, Miller EA, Goldberg J, Orci L, Schekman R . Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol 2004; 20:87–123.
Rabouille C, Klumperman J . Opinion: The maturing role of COPI vesicles in intra-Golgi transport. Nat Rev Mol Cell Biol 2005; 6:812–817.
Cornell CT, Kiosses WB, Harkins S, Whitton JL . Inhibition of protein trafficking by coxsackievirus b3: multiple viral proteins target a single organelle. J Virol 2006; 80:6637–6647.
Doedens JR, Kirkegaard K . Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J 1995; 14:894–907.
Hsu NY, Ilnytska O, Belov G, et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010; 141:799–811.
Wessels E, Duijsings D, Notebaart RA, Melchers WJ, van Kuppeveld FJ . A proline-rich region in the coxsackievirus 3A protein is required for the protein to inhibit endoplasmic reticulum-to-golgi transport. J Virol 2005; 79:5163–5173.
Belov GA, Feng Q, Nikovics K, Jackson CL, Ehrenfeld E . A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog 2008; 4:e1000216.
Wessels E, Duijsings D, Niu TK, et al. A viral protein that blocks Arf1-mediated COP-I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev Cell 2006; 11:191–201.
D'Angelo G, Vicinanza M, Di Campli A, De Matteis MA . The multiple roles of PtdIns(4)P – not just the precursor of PtdIns(4,5)P2. J Cell Sci 2008; 121:1955–1963.
Graham TR, Burd CG . Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol 2011; 21:113–121.
Santiago-Tirado FH, Bretscher A . Membrane-trafficking sorting hubs: cooperation between PI4P and small GTPases at the trans-Golgi network. Trends Cell Biol 2011; 21:515–525.
Blumental-Perry A, Haney CJ, Weixel KM, Watkins SC, Weisz OA, Aridor M . Phosphatidylinositol 4-phosphate formation at ER exit sites regulates ER export. Dev Cell 2006; 11:671–682.
Lorente-Rodriguez A, Barlowe C . Requirement for Golgi-localized PI(4)P in fusion of COPII vesicles with Golgi compartments. Mol Biol Cell 2011; 22:216–229.
Arita M, Wakita T, Shimizu H . Characterization of pharmacologically active compounds that inhibit poliovirus and enterovirus 71 infectivity. J Gen Virol 2008; 89:2518–2530.
Balla A, Tuymetova G, Toth B, et al. Design of drug-resistant alleles of type-III phosphatidylinositol 4-kinases using mutagenesis and molecular modeling. Biochemistry 2008; 47:1599–1607.
Knight ZA, Gonzalez B, Feldman ME, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 2006; 125:733–747.
Arita M, Kojima H, Nagano T, Okabe T, Wakita T, Shimizu H . Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J Virol 2011; 85:2364–2372.
Balla A, Tuymetova G, Tsiomenko A, Varnai P, Balla T . A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-III alpha: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol Biol Cell 2005; 16:1282–1295.
Godi A, Di Campli A, Konstantakopoulos A, et al. FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat Cell Biol 2004; 6:393–404.
He J, Scott JL, Heroux A, et al. Molecular basis of phosphatidylinositol 4-phosphate and ARF1 GTPase recognition by the FAPP1 pleckstrin homology (PH) domain. J Biol Chem 2011; 286:18650–18657.
Toth B, Balla A, Ma H, Knight ZA, Shokat KM, Balla T . Phosphatidylinositol 4-kinase IIIbeta regulates the transport of ceramide between the endoplasmic reticulum and Golgi. J Biol Chem 2006; 281:36369–36377.
Godi A, Pertile P, Meyers R, et al. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol 1999; 1:280–287.
Kahn RA, Volpicelli-Daley L, Bowzard B, et al. Arf family GTPases: roles in membrane traffic and microtubule dynamics. Biochem Soc Trans 2005; 33:1269–1272.
Hausser A, Storz P, Martens S, Link G, Toker A, Pfizenmaier K . Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase IIIbeta at the Golgi complex. Nat Cell Biol 2005; 7:880–886.
Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J 2007; 408:297–315.
Lackey K, Cory M, Davis R, et al. The discovery of potent cRaf1 kinase inhibitors. Bioorg Med Chem Lett 2000; 10:223–226.
Prudent R, Moucadel V, Lopez-Ramos M, et al. Expanding the chemical diversity of CK2 inhibitors. Mol Cell Biochem 2008; 316:71–85.
Horiuchi KY, Wang Y, Diamond SL, Ma H . Microarrays for the functional analysis of the chemical-kinase interactome. J Biomol Screen 2006; 11:48–56.
Li M, Smith CJ, Walker MT, Smith TJ . Novel inhibitors complexed with glutamate dehydrogenase: allosteric regulation by control of protein dynamics. J Biol Chem 2009; 284:22988–23000.
Levine TP and Munro S . Targeting of Golgi-specific pleckstrin homology domains involves both PtdIns 4-kinase-dependent and -independent components. Curr Biol 2002; 12:695–704.
Berger KL, Cooper JD, Heaton NS, et al. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci USA 2009; 106:7577–7582.
Berger KL, Kelly SM, Jordan TX, Tartell MA, Randall G . Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J Virol 2011; 85:8870–8883.
Borawski J, Troke P, Puyang X, et al. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J Virol 2009; 83:10058–10074.
Lim YS, Hwang SB . Hepatitis C virus NS5A protein interacts with phosphatidylinositol 4-kinase type IIIalpha and regulates viral propagation. J Biol Chem 2011; 286:11290–11298.
Reiss S, Rebhan I, Backes P, et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011; 9:32–45.
Tai AW, Benita Y, Peng LF, et al. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009; 5:298–307.
Tai AW, Salloum S . The role of the phosphatidylinositol 4-kinase PI4KA in hepatitis C virus-induced host membrane rearrangement. PLoS One 2011; 6:e26300.
Trotard M, Lepere-Douard C, Regeard M, et al. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J 2009; 23:3780–3789.
Mackenzie JM, Khromykh AA, Parton RG . Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe 2007; 2:229–239.
Martin-Acebes MA, Blazquez AB, Jimenez de ON, Escribano-Romero E, Saiz JC . West Nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PLoS One 2011; 6:e24970.
Heaton NS, Perera R, Berger KL, et al. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc Natl Acad Sci USA 2010; 107:17345–17350.
Perera R, Riley C, Isaac G, et al. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog 2012; 8:e1002584.
Heaton NS, Randall G . Multifaceted roles for lipids in viral infection. Trends Microbiol 2011; 19:368–375.
Crotty S, Saleh MC, Gitlin L, Beske O, Andino R . The poliovirus replication machinery can escape inhibition by an antiviral drug that targets a host cell protein. J Virol 2004; 78:3378–3386.
Belov GA, Kovtunovych G, Jackson CL, Ehrenfeld E . Poliovirus replication requires the N-terminus but not the catalytic Sec7 domain of ArfGEF GBF1. Cell Microbiol 2010; 12:1463–1479.
Coelmont L, Hanoulle X, Chatterji U, et al. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 2010; 5:e13687.
Lanke KH, van der Schaar HM, Belov GA, et al. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J Virol 2009; 83:11940–11949.
Reed LJ, Muench H . A simple method of estimating fifty percent endpoints. Am J Hyg 1938; 27:493–497.
Zhao XH, Bondeva T, Balla T . Characterization of recombinant phosphatidylinositol 4-kinase beta reveals auto- and heterophosphorylation of the enzyme. J Biol Chem 2000; 275:14642–14648.
Jung G, Barylko B, Lu D, Shu H, Yin H, Albanesi JP . Stabilization of phosphatidylinositol 4-kinase type IIbeta by interaction with Hsp90. J Biol Chem 2011; 286:12775–12784.
van Kuppeveld FJ, Galama JM, Zoll J, Melchers WJ . Genetic analysis of a hydrophobic domain of coxsackie B3 virus protein 2B: a moderate degree of hydrophobicity is required for a cis-acting function in viral RNA synthesis. J Virol 1995; 69:7782–7790.
Acknowledgements
We are grateful to Drs K Conrath and M Andrews (Galapagos NV, Belgium) for performing the in vitro PI4K activity assay. We also thank Drs N Altan-Bonnet (Rutgers University, USA) and T Balla (National Institute for Health, USA) for fruitful discussions and for providing plasmids, Dr F Wieland (Biochemie-Zentrum, Germany) for providing antibodies and Dr J Albanesi (UT Southwestern Medical Center, Texas) for providing plasmids. This work was supported by research grants from the “Covenant K.U. Leuven-Radboud University Nijmegen” framework, the Netherlands Organization for Scientific Research (NWO-ECHO-700.57.001 and NWO-ALW-820.02.018), and the SILVER Large Scale Collaborative Project (grant agreement number 260644) of the European Union 7th Framework. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Golgi-localization of FAPP1-PH-GFP is predominantly determined by PI4KIIIβ. (PDF 57 kb)
Supplementary information, Figure S2
Enviroxime, GW5074, and PIK93 do not affect PI4KIIIβ recruitment by wild-type 3A or 3A-H57Y. (PDF 118 kb)
Supplementary information, Figure S3
SiRNA-treatment does not induce cytotoxic effects on cells. (PDF 44 kb)
Supplementary information, Figure S4
Efficient depletion of PI4KIIβ by siRNA-treatment. (PDF 43 kb)
Rights and permissions
About this article
Cite this article
van der Schaar, H., van der Linden, L., Lanke, K. et al. Coxsackievirus mutants that can bypass host factor PI4KIIIβ and the need for high levels of PI4P lipids for replication. Cell Res 22, 1576–1592 (2012). https://doi.org/10.1038/cr.2012.129
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2012.129
Keywords
This article is cited by
-
Human-type sialic acid receptors contribute to avian influenza A virus binding and entry by hetero-multivalent interactions
Nature Communications (2022)
-
Novel capsid binder and PI4KIIIbeta inhibitors for EV-A71 replication inhibition
Scientific Reports (2021)
-
The Golgi protein ACBD3 facilitates Enterovirus 71 replication by interacting with 3A
Scientific Reports (2017)
-
A new paradigm in viral resistance
Cell Research (2012)
