
Abstract
The extracellular signal-regulated kinase 1/2 (ERK1/2) cascade is the prototype mammalian mitogen-activated protein kinase (MAPK) signaling cascade that regulates a number of processes, including proliferation, differentiation, survival, migration, stress responses and apoptosis. How this seemingly linear cascade is modulated to achieve a specific cellular function has been a main focus of the field. In this review, we describe new as well as old findings in the regulation of the ERK1/2 pathway in normal and disease states via MAP3Ks.
Similar content being viewed by others


Introduction
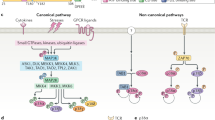
Since we first cloned the extracellular signal-regulated kinases 1/2 (ERK1/2) over twenty-one years ago, a myriad of growth factors and cytokines have been identified as their activators1. Originally described as microtubule-associated protein kinases activated by insulin, and now known as mitogen-activated protein kinases (MAPK), these enzymes are regulated through a protein kinase cascade (Figure 1). The cascade is usually initiated by ligands that bind a variety of membrane receptors, which engage adaptor proteins and exchange factors to induce activation of the small G protein, Ras2,3,4,5,6,7. GTP-bound active Ras then recruits and activates the first protein kinase in the cascade, one or more of the Raf proteins, Raf-1 (C-Raf), B-Raf, and/or A-Raf8,9. Raf kinases phosphorylate and activate the MAP2K protein kinases, MEK1/2, that eventually phosphorylate and activate ERK1/210,11,12,13. While this seemingly simple, linear cascade involves progressive signal amplification initiated by ligands and governed by phosphorylation, proteins associated with other pathways and an array of modifications fine tune the signal. Integration of these other inputs directs the MAPK cascade to couple signals from specific stimuli to generate subsequent cellular activities that may lead to differentiation, proliferation, and motility, for example, as appropriate14. Understanding how cells interpret and process abundant and diverse inputs remains challenging. Cells use scaffolding proteins, phosphatases, and second messengers to refine complex signaling programs connecting extracellular stimuli to intracellular responses. This review will focus on understanding how such proteins control signaling at the MAP3K level to regulate attenuation or augmentation of the ERK1/2 pathway.

Activity of the ERK1/2 MAPK cascade can be modulated by inputs at different sites in the pathway. The best studied mechanism of ERK1/2 activation is through receptor tyrosine kinases. Signaling events at the plasma membrane initiate the cascade through ligand/receptor binding. The signal is transmitted by recruiting adaptor proteins (e.g., Grb2) and exchange factors (e.g., SOS) that induce the activation of Ras at the plasma membrane. The activated, GTP-bound Ras then transmits the signal by activating Raf protein kinases (MAP3Ks). Rafs activate MEK1/2 through phosphorylation, which in turn phosphorylate ERK1/2. Once activated, ERK1/2 propagate the signal by affecting a variety of downstream signaling pathways. Additional components that can provide inputs to the cascade are shown including a KSR-associated AKAP and the calcium-dependent phosphatase calcineurin.
B-Raf in cancer
Homologs of all three Raf serine/threonine protein kinase family members are found in vertebrates, while a single Raf equivalent exists in invertebrates15. The Raf proteins contain three conserved regions, CR1, CR2, and CR3. CR1 includes the Ras-binding domain; CR3 comprises the kinase domain, which is typical of the protein kinase family, containing two folding domains with adenosine triphosphate-binding site at their interface and a regulatory activation loop at the mouth of the active site. B-Raf is the most commonly mutated protein kinase in the human genome; most of the mutations occur in the kinase domain, either in the glycine-rich loop or the activation loop16,17 (Table 1). A substitution of glutamate for valine at residue 600 accounts for 90 % of the B-Raf mutations observed in human cancers including, malignant melanoma, colorectal cancer, ovarian, and papillary thyroid carcinomas. In addition to V600E, > 30 other mutations of the BRAF gene have been found in human cancers, the most frequent of these mutations in melanoma17,18. The frequency of mutations in B-Raf and not A- or C-Raf may be attributed to melanocyte biology as well as to details of its constitutive activation and regulation.
B-Raf/C-Raf interactions in cancer and cancer therapeutics
Phosphorylation of the activation loop is required for the activity of all three Raf family members. In addition to having a higher basal kinase activity than C-Raf or A-Raf, B-Raf also lacks the need for tyrosine or multiple serine phosphorylations that are required for the activity of A-Raf and C-Raf19. Higher B-Raf activity may be due to Ras-independent constitutive phosphorylation of S445, in a region preceding the catalytic domain. Activation of the pathway can also be attributed to the formation of Ras-dependent homo- and heterodimers, between B-Raf and C-Raf9,20. A much greater understanding of the relevance of B-Raf/C-Raf interactions has arisen from the analysis of the mechanisms of action of second-generation selective oncogenic B-Raf inhibitors. Several of these compounds, while initially inhibiting ERK1/2 phosphorylation, eventually promote C-Raf/ERK1/2 activation, leading to an enhanced cancer cell proliferation or drug refractory tumors21,22,23,24. Insights into how several of these inhibitors become activators of the MAPK pathway ultimately revealed that pathway activation occurred from drug-induced dimerization of C-Raf with wild type or mutant B-Raf. Even kinase dead B-Raf (a mimic of inhibited B-Raf) promotes signaling22,23,25. The strategy underlying the development of second-generation inhibitors was to target mutant B-Raf selectively. Thus, such inhibitors had no inhibitory effect on cells with other activated oncogenes, such as mutant Ras, if they have wild-type B-Raf22,23,25. In addition to mechanisms involving C-Raf, several of the Raf inhibitors also induced B-Raf interaction with the kinase suppressor of Ras 1 (KSR1, discussed further below) that competed with C-Raf to attenuate the signaling pathway26. B-Raf complexes formed with KSR1 differ greatly from the complexes that induce C-Raf binding. In addition to altering the activating effects observed during signaling to ERK1/2, the KSR/B-Raf complexes are also Ras-independent, suggesting that KSR1 expression may be important for inhibitor efficacy.
Kinase suppressor of Ras
Originally discovered in model organisms as a protein required for activated Ras to accelerate MAPK signaling, KSR isoforms are members of the protein kinase superfamily structurally related to Raf. The relevance of the catalytic activity of KSR proteins is in debate27,28,29,30,31. KSRs contain arginine instead of the invariant catalytic lysine in the N-terminal region of the kinase domain present in other protein kinases. KSR1 may be most important as a scaffold due to its ability to bind simultaneously to components at each level of the MAPK cascade (MAP3K, MAP2K, and MAPK). Because signaling is diminished if it is expressed in excess of its binding partners, KSR1 fits the characteristics of a scaffold27,30,32. A functional catalytic lysine appears to be absent from KSR, unlike what we found in WNK protein kinases; in WNKs the catalytic lysine is not in the canonical position, but it is nearby in a distinct structural element33. Nevertheless, KSR1 purified from E. coli can directly phosphorylate MEK1 in vitro, albeit at a rate that may be representative of a catalytically impaired Raf mutant29. Regardless, KSR is a critical component of the MAPK pathway and thus subject to modulation just as are the other proteins in the pathway. The E3 ligase and Ras effector protein, impedes mitogenic signal propagation (IMP), attenuates Raf-MEK association via inactivation of KSR homo- and heterodimerization with RAF proteins34. The association between IMP and KSR1 also blocks KSR1 interaction with MEK1, preventing activation of ERK1/235.
While Drosophila and C. elegans have only one KSR protein, mammals have two KSR family members27. KSR1 knockout mice have been generated and show no extreme abnormalities other than modestly impaired immune and neuronal signaling, suggesting a functional redundancy between KSR1 and KSR236,37. KSR2 shares 43% sequence identity with KSR1, which includes the conserved regions required to bind to the three members of the ERK1/2 cascade, and maintains the ability to regulate pathway activation in a concentration-dependent manner32. However, novel roles for KSR2 have been reported in metabolic pathways. KSR2 has been shown to regulate AMPK to promote glucose and fatty acid metabolism, and its loss in mouse models leads to obesity and insulin resistance38.
Glucose metabolism and MAPK activation
Pancreatic β cells are specialized endocrine cells that secrete insulin in response to elevated blood glucose in order to facilitate proper glucose utilization. Glucose metabolism in β cells causes an increase in the adenosine triphosphate/adenosine diphosphate ratio, which leads to closure of the adenosine triphosphate-dependent potassium channel, ultimately resulting in calcium entry into the cells and release of insulin. Thus, fluctuations in intracellular calcium are integral to β-cell function. ERK1 and ERK2 have significant roles in maintaining β cell function. We find that the nutrient-dependent regulation of ERK1/2 and their nutrient-dependent actions are also controlled by calcium39. In β cells, ERK1/2 are activated by both glucose and other nutrients, as well as other factors that potentiate insulin secretion such as glucagon-like peptide, which acts through the second messenger cAMP39.
Glucose-induced insulin gene transcription has been shown to require a set of core transcription factors, including BETA2 (also known as NeuroD1), PDX1, MafA, and NFAT29,40,41,42,43,44,45,46. We showed that these transcription factors act synergistically to promote insulin gene transcription in an ERK1/2-dependent manner, as phosphorylation of both Beta2 and PDX-1 is direct47. Moreover, transactivation of NFAT and MafA is also ERK1/2-sensitive, as inhibition of the pathway via the MEK1/2 inhibitor U0126 dissociates both factors from the insulin promoter40.
Previous work in the immune system showed that the calcium- and calmodulin-dependent phosphatase calcineurin is required for NFAT function48. Consistent with their actions in the immune system, calcineurin inhibitors, such as FK506 and cyclosporin A, are immunosuppressants that decrease the rejection of transplanted tissues, including pancreatic islets. These inhibitors, however, reduce insulin gene transcription and secretion, which may further exacerbate complications in diabetic patients following islet transplantation40,49.
Calcium-dependent phosphatases
In addition to phosphoprotein phosphatases 1 and 2A and the canonical dual-specificity phosphatases that modulate the distal steps in the MAPK pathway, calcineurin also has a critical role in control of ERK1/2 in pancreatic β cells. Glucose metabolism and the increases in second messengers, noted above, increase cytosolic calcium by inducing calcium entry and/or its release from intracellular stores39,50. We found that increased cytosolic calcium in β cells directs the calcineurin-dependent activation of ERK1/2 that can be pharmacologically blocked with calcineurin inhibitors, e.g., cyclosporin A, FK506, and its analog FK52039 (see Figure 2 and below). Calcineurin knockout mice have been generated. Mice with β-cell-specific deletion of the calcineurin regulatory subunit, calcineurin b1, were found to develop diabetes that is distinguished by decreased β-cell proliferation and mass as well as reduced pancreatic insulin content51. Interestingly, these defects could be rescued by expression of active NFAT. Attempts to understand the mechanism by which calcineurin activates MAPK kinase cascades have yielded interesting and perhaps unexpected outcomes.

Calcineurin reverses negative feedback on B-Raf to permit ERK1/2 activation by nutrients in pancreatic β cells. Once activated, ERK1/2 can phosphorylate multiple upstream components in the pathway. In β cells, in response to nutrients ERK1/2 phosphorylate B-Raf preventing its continued stimulation of MEK1/2. B-Raf is required for the activation of ERK1/2 by nutrients. The calcium-dependent phosphatase calcineurin can dephosphorylate a key inhibitory site on B-Raf, allowing continued ERK1/2 activation.
In both a pancreatic β cell line and a neuroblastoma cell line KSR2 but not KSR1, is a calcineurin substrate32. Activity of the phosphatase contributes to the localization of KSR2 to the membrane, where it can activate the ERK1/2 pathway32,39. Inhibition of calcineurin activity under these conditions reduces activation of ERK1/2. This regulation is calcium-dependent, providing mechanistic insight into activation of ERK1/2 by calcium. We had previously shown that dominant negative mutants of both Ras and Raf inhibited glucose-dependent ERK1/2 activation in pancreatic β cells, which is consistent with the idea that both are required for glucose-dependent ERK1/2 activation39. Importantly, we found that in pancreatic β cells both B-Raf activity and B-Raf/C-Raf dimerization are enhanced in a calcineurin- and glucose-dependent manner52.
Negative feedback
Positive and negative feedback loops have critical roles in modulation of signal transduction cascades. Growth factors that activate the MAPK pathway have been suggested to regulate the MAPK phosphatases that turn off the kinases in the same pathways53,54. Expression of B-Raf, but not C-Raf, in 293 cells led to increased ERK1/2 activation; however, B-Raf is subject to ERK1/2-mediated feedback inhibition as we found that treatment with the MEK inhibitor, U0126, in the absence of calcineurin led to increased B-Raf activity52 (Figure 2). This suggests that calcineurin promotes B-Raf activation by interrupting the negative feedback effects of ERK1/2. ERK1/2 have been shown to phosphorylate B-Raf on residues S151, S750, T401, and T753, to disrupt not only active Ras interaction but also homo- and heterodimer formation55. Phosphorylation of T401 by ERK1/2 is reduced by U0126 and can specifically be removed by calcineurin to promote B-Raf activation and interaction with C-Raf52.
ERK2 is fully activated by dual phosphorylation of both threonine-183 and tyrosine-185 residues56,57. Canonical MAPK phosphatases or dual-specificity (DUSP) enzymes dephosphorylate both phosphothreonine and phosphotyrosine residues within ERK1 and ERK258,59. In addition to the various extracellular stimuli that activate MAPK phosphatase-1, the second messenger cAMP has also been shown to stimulate activation of MAPK phosphatase-160. The ability of the cAMP pathway to inhibit growth of tumors transformed by activation of MAPK was a topic of much interest for many years61. Later it was discovered that there is a cross talk between cAMP and MAPK pathways, and the effects were differentially depending on cell type and context.
Cross talk of cAMP and MAPK pathways
In cells such as melanocytes, thyroid cells, cells of the anterior pituitary, and β cells, cAMP can enhance proliferation or differentiation; however, in the eye, lung, certain fibroblasts, astrocytes, and hepatocytes to name a few cAMP decreases proliferation39,61,62,63,64,65,66. Activation of MAPK pathway by cAMP occurs in cells that are neural crest-derived, such as melanocytes and the neuroendocrine pheochromocytoma PC12 cell line67,68.
Effectors of cAMP include the multifunctional cAMP-dependent protein kinase (PKA) as well as the guanine nucleotide exchange factors, Epac1 and 2. Epacs are exchange factors for the small G proteins Ras-related protein 1 and 2 (Rap1, Rap 2)69. Activation of ERK1/2 by cAMP that induces proliferative signals is Ras- and B-Raf-mediated in melanocytes, whereas the differentiation program may solely rely on Epac activation70,71. Proper signaling in the cAMP pathway is also achieved by compartmentalization of PKA and Epacs by the scaffolding proteins known as A-kinase anchoring proteins. Our laboratory identified the only AKAP shown to bind directly to ERK1/2, radial spoke protein 3 (RSPH3) (Figure 3). RSPH3 was identified in a yeast two-hybrid screen and it binds to ERK1/272. ERK1/2 phosphorylates the anchoring protein; this phosphorylation regulates the ability of RSPH3 to bind to the regulatory subunits of PKA. Inhibition of ERK1/2 activity in this setting negatively regulates the AKAP function of RSPH3. Several A-kinase anchoring proteins have been shown to mediate cross talk between PKA and the MAPK pathway by interacting with other components of the cascade. Recently AKAP-Lbc has been shown to amplify the positive effect that cAMP has on the MAPK pathway via its interaction with KSR1. In addition to its known function as a guanine nucleotide exchange factors, AKAP-Lbc is also a scaffold protein73. AKAP-Lbc binds to KSR1, which facilitates cAMP-dependent phosphorylation of KSR1 by PKA, ensuring sustained activation of ERK1/274. While these findings support the idea that PKA has a positive function in ERK1/2 activation, this is not always the case.

RSPH3 3 is a mammalian AKAP that binds ERK1/2. RSPH3 is the human homolog of a protein found in motile cilia that forms part of radial spokes that connect the inner to the outer pairs of microtubules. RSPH3 is also an AKAP. ERK1/2 bind directly to RSPH3 and phosphorylate it, altering its interactions with the regulatory subunit of PKA.
As mentioned above, cAMP activation of ERK1/2 that leads to proliferation in melanocytes is mediated by B-Raf. At the same time, C-Raf is inhibited by PKA via phosphorylation on several sites. Mechanistically, these phosphorylations both inhibit its interaction with Ras and alter the binding with the adaptor protein 14-3-375,76. Ras-dependent activation of C-Raf requires not only activation loop phosphorylation but also proper binding and dimerization of 14-3-377,78. In melanomas, Ras mutations and B-Raf mutations are usually mutually exclusive. If Ras is mutated, the pathway switches from B-Raf to C-Raf to activate ERK1/267. Overexpression of activated N-Ras in melanocytes hyperactivates ERK1/2, causing phosphorylation of S151 on B-Raf. This promotes the switch to C-Raf and melanocyte transformation79. In this context, cAMP is degraded by phosphodiesterases (PDE)79. Two isoforms of PDE4 are upregulated in N-Ras-transformed melanocytes, and are activated by ERK1/2. Loss of PDE4B2 reduces the ability of N-Ras to transform melanocytes, suggesting the requirement for PDE4 in the process79.
Even though cAMP has been shown to inhibit proliferation, the inhibition is not ERK1/2-dependent. The ΔRaf1:ER fusion protein lacks amino terminal residues and is thus resistant to cAMP. ERK1/2, activated in this context, are not sensitive to elevated cAMP, although DNA synthesis, CDK2 activity, and cyclin A expression are80. In addition to the inhibitory effects of cAMP on ERK1/2, we find that ERK5 is also uncoupled from activating signals in a PKA-dependent manner81.
MAPK pathway in other cancers
Historically aberrant activation of the ERK1/2 pathway either by mutations found in receptor tyrosine kinases or Ras or Raf, have been shown to lead to increased survival and/or proliferation of malignant cells. This general view is challenged by the behavior of small cell lung cancer (SCLC). Lung cancer is usually classified as SCLC or non-small cell lung cancer (NSCLC). Patients with SCLC often have metastases at the time of diagnosis, leading to a very poor long-term prognosis82. These two types of lung cancer are characterized by the differences in mutation profile. Activating mutations in the ERK1/2 pathway are found frequently in NSCLC and almost never in SCLC83. In contrast to most of the systems discussed above, in SCLC activated Raf-1 causes growth arrest84. ERK1/2 can also be activated by receptor tyrosine kinases, such as c-Met and its ligand hepatocyte growth factor, and cKit and its ligand stem cell factor, which establishes an autocrine loop in SCLC85,86,87. cKit and stem cell factor are expressed frequently in SCLC, but not in NSCLC. Inhibition of MEK1/2 does not impair growth of SCLC. This may, in part, be due to the presence of mutant retinoblastoma protein 1, as expression of wild-type retinoblastoma protein 1 can sensitize these cells to the MEK1/2 inhibitor87. Thus, it appears that MEK inhibitor insensitivity in SCLC arises from the loss of retinoblastoma protein 1 function. Loss or mutation of retinoblastoma protein 1 and p53 in pulmonary neuroendocrine cells is sufficient to cause a mouse model of SCLC88,89. When various isoforms of PI 3-kinase are overexpressed in SCLC they increase stem cell factor-stimulated activation of AKT, but not ERK1/286. Additionally, an analysis of 42 patients with SCLC revealed that while about half were found to have active ERK1/2, AKT and c-Kit, the active ERK was cytoplasmic, not nuclear, and correlated positively with patient survival90.
Conclusion
The ERK1/2 signaling cascade is central to regulation of many, sometimes opposing, cellular functions. Here, we point to new findings that determine signaling specificity in tissue-dependent contexts in both normal and pathological states. Indeed, dysregulation of the ERK1/2 signaling cascade has been noted in neurodegenerative disease91 and diabetes,92,93 as well as in cancer. The effective targeting of this pathway in disease relies upon the understanding of intricacies in modulating all of the signaling components. Additionally, elucidating cross talk of MAPK components with other signaling pathways that induce and promote pathologies could also provide new ground for combination therapies.
References
Boulton TG, Yancopoulos GD, Gregory JS, et al. An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science 1990; 249:64–67.
Robinson LC, Gibbs JB, Marshall MS, Sigal IS, Tatchell K . CDC25: a component of the RAS-adenylate cyclase pathway in Saccharomyces cerevisiae. Science 1987; 235:1218–1221.
Broek D, Toda T, Michaeli T, et al. The S. cerevisiae CDC25 gene product regulates the RAS/adenylate cyclase pathway. Cell 1987; 48:789–799.
Simon MA, Bowtell DD, Dodson GS, Laverty TR, Rubin GM . Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell 1991; 67:701–716.
Olivier JP, Raabe T, Henkemeyer M, et al. A Drosophila SH2-SH3 adaptor protein implicated in coupling the sevenless tyrosine kinase to an activator of Ras guanine nucleotide exchange. Sos. Cell 1993; 73:179–191.
Lowenstein EJ, Daly RJ, Batzer AG, et al. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992; 70:431–442.
Rogge RD, Karlovich CA, Banerjee U . Genetic dissection of a neurodevelopmental pathway: Son of sevenless functions downstream of the sevenless and EGF receptor tyrosine kinases. Cell 1991; 64:39–48.
Van Aelst L, Barr M, Marcus S, Polverino A, Wigler M . Complex formation between RAS and RAF and other protein kinases. Proc Natl Acad Sci USA 1993; 90:6213–6217.
Luo Z, Tzivion G, Belshaw PJ, Vavvas D, Marshall M, Avruch J . Oligomerization activates c-Raf-1 through a Ras-dependent mechanism. Nature 1996; 383:181–185.
Dent P, Haser W, Haystead TA, Vincent LA, Roberts TM, Sturgill TW . Activation of mitogen-activated protein kinase kinase by v-Raf in NIH 3T3 cells and in vitro. Science 1992; 257:1404–1407.
Kyriakis JM, App H, Zhang XF, et al. Raf-1 activates MAP kinase-kinase. Nature 1992; 358:417–421.
Seger R, Ahn NG, Posada J, et al. Purification and characterization of mitogen-activated protein kinase activator(s) from epidermal growth factor-stimulated A431 cells. J Biol Chem 1992; 267:14373–14381.
Robbins DJ, Cobb MH . Extracellular signal-regulated kinases 2 autophosphorylates on a subset of peptides phosphorylated in intact cells in response to insulin and nerve growth factor: analysis by peptide mapping. Mol Biol Cell 1992; 3:299–308.
Marshall CJ . Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 1995; 80:179–185.
Michaloglou C, Vredeveld LC, Mooi WJ, Peeper DS . BRAF(E600) in benign and malignant human tumours. Oncogene 2008; 27:877–895.
Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007; 446:153–158.
Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004; 116:855–867.
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949–954.
Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R . Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J 1999; 18:2137–2148.
Weber CK, Slupsky JR, Kalmes HA, Rapp UR . Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res 2001; 61:3595–3598.
Emuss V, Garnett M, Mason C, Marais R . Mutations of C-RAF are rare in human cancer because C-RAF has a low basal kinase activity compared with B-RAF. Cancer Res 2005; 65:9719–9726.
Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010; 464:431–435.
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N . RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010; 464:427–430.
Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA 2008; 105:3041–3046.
Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010; 140:209–221.
McKay MM, Ritt DA, Morrison DK . RAF inhibitor-induced KSR1/B-RAF binding and its effects on ERK cascade signaling. Curr Biol 2011; 21:563–568.
Therrien M, Chang HC, Solomon NM, Karim FD, Wassarman DA, Rubin GM . KSR, a novel protein kinase required for RAS signal transduction. Cell 1995; 83:879–888.
Therrien M, Michaud NR, Rubin GM, Morrison DK . KSR modulates signal propagation within the MAPK cascade. Genes Dev 1996; 10:2684–2695.
Goettel JA, Liang D, Hilliard VC, et al. KSR1 is a functional protein kinase capable of serine autophosphorylation and direct phosphorylation of MEK1. Exp Cell Res 2011; 317:452–463.
Roy F, Laberge G, Douziech M, Ferland-McCollough D, Therrien M . KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev 2002; 16:427–438.
Sundaram M, Han M . The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell 1995; 83:889–901.
Dougherty MK, Ritt DA, Zhou M, et al. KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol Cell 2009; 34:652–662.
Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH . WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem 2000; 275:16795–16801.
Matheny SA, Chen C, Kortum RL, Razidlo GL, Lewis RE, White MA . Ras regulates assembly of mitogenic signalling complexes through the effector protein IMP. Nature 2004; 427:256–260.
Chen C, Lewis RE, White MA . IMP modulates KSR1-dependent multivalent complex formation to specify ERK1/2 pathway activation and response thresholds. J Biol Chem 2008; 283:12789–12796.
Nguyen A, Burack WR, Stock JL, et al. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol Cell Biol 2002; 22:3035–3045.
Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD . Kinase suppressor of Ras1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron 2006; 50:765–779.
Costanzo-Garvey DL, Pfluger PT, Dougherty MK, et al. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab 2009; 10:366–378.
Arnette D, Gibson TB, Lawrence MC, et al. Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic beta cells. J Biol Chem 2003; 278:32517–32525.
Lawrence MC, McGlynn K, Park BH, Cobb MH . ERK1/2-dependent activation of transcription factors required for acute and chronic effects of glucose on the insulin gene promoter. J Biol Chem 2005; 280:26751–26759.
Marshak S, Totary H, Cerasi E, Melloul D . Purification of the beta-cell glucose-sensitive factor that transactivates the insulin gene differentially in normal and transformed islet cells. Proc Natl Acad Sci USA 1996; 93:15057–15062.
Naya FJ, Stellrecht CM, Tsai MJ . Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev 1995; 9:1009–1019.
Lawrence MC, Bhatt HS, Easom RA . NFAT regulates insulin gene promoter activity in response to synergistic pathways induced by glucose and glucagon-like peptide-1. Diabetes 2002; 51:691–698.
Lawrence MC, Bhatt HS, Watterson JM, Easom RA . Regulation of insulin gene transcription by a Ca(2+)-responsive pathway involving calcineurin and nuclear factor of activated T cells. Mol Endocrinol 2001; 15:1758–1767.
Sander M, Griffen SC, Huang J, German MS . A novel glucose-responsive element in the human insulin gene functions uniquely in primary cultured islets. Proc Natl Acad Sci USA 1998; 95:11572–11577.
German MS, Wang J . The insulin gene contains multiple transcriptional elements that respond to glucose. Mol Cell Biol 1994; 14:4067–4075.
Khoo S, Griffen SC, Xia Y, Baer RJ, German MS, Cobb MH . Regulation of insulin gene transcription by ERK1 and ERK2 in pancreatic beta cells. J Biol Chem 2003; 278:32969–32977.
Luo C, Burgeon E, Carew JA, et al. Recombinant NFAT1 (NFATp) is regulated by calcineurin in T cells and mediates transcription of several cytokine genes. Mol Cell Biol 1996; 16:3955–3966.
Robertson RP . Cyclosporin-induced inhibition of insulin secretion in isolated rat islets and HIT cells. Diabetes 1986; 35:1016–1019.
Holz GG, Leech CA, Heller RS, Castonguay M, Habener JF . cAMP-dependent mobilization of intracellular Ca2+ stores by activation of ryanodine receptors in pancreatic beta-cells. A Ca2+ signaling system stimulated by the insulinotropic hormone glucagon-like peptide-1-(7-37). J Biol Chem 1999; 274:14147–14156.
Heit JJ, Apelqvist AA, Gu X, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature 2006; 443:345–349.
Duan L, Cobb MH . Calcineurin increases glucose activation of ERK1/2 by reversing negative feedback. Proc Natl Acad Sci USA 2010; 107:22314–22319.
Bokemeyer D, Sorokin A, Yan M, Ahn NG, Templeton DJ, Dunn MJ . Induction of mitogen-activated protein kinase phosphatase 1 by the stress-activated protein kinase signaling pathway but not by extracellular signal-regulated kinase in fibroblasts. J Biol Chem 1996; 271:639–642.
Beltman J, McCormick F, Cook SJ . The selective protein kinase C inhibitor, Ro-31-8220, inhibits mitogen-activated protein kinase phosphatase-1 (MKP-1) expression, induces c-Jun expression, and activates Jun N-terminal kinase. J Biol Chem 1996; 271:27018–27024.
Ritt DA, Monson DM, Specht SI, Morrison DK . Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol 2010; 30:806–819.
Seger R, Ahn NG, Posada J, et al. Purification and characterization of mitogen-activated protein kinase activator(s) from epidermal growth factor-stimulated A431 cells. J Biol Chem 1992; 267:14373–14381.
Anderson NG, Maller JL, Tonks NK, Sturgill TW . Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature 1990; 343:651–653.
Alessi DR, Smythe C, Keyse SM . The human CL100 gene encodes a Tyr/Thr-protein phosphatase which potently and specifically inactivates MAP kinase and suppresses its activation by oncogenic ras in Xenopus oocyte extracts. Oncogene 1993; 8:2015–2020.
Camps M, Nichols A, Gillieron C, et al. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science 1998; 280:1262–1265.
Brion L, Maloberti PM, Gomez NV, et al. MAPK phosphatase-1 (MKP-1) expression is up-regulated by hCG/cAMP and modulates steroidogenesis in MA-10 Leydig cells. Endocrinology 2011; 152:2665–2677.
Ally S, Clair T, Katsaros D, et al. Inhibition of growth and modulation of gene expression in human lung carcinoma in athymic mice by site-selective 8-Cl-cyclic adenosine monophosphate. Cancer Res 1989; 49:5650–5655.
Cook SJ, McCormick F . Inhibition by cAMP of Ras-dependent activation of Raf. Science 1993; 262:1069–1072.
Gagelin C, Toru-Delbauffe D, Gavaret JM, Pierre M . Effects of cyclic AMP on components of the cell cycle machinery regulating DNA synthesis in cultured astrocytes. J Neurochem 1999; 73:1799–1805.
Kikukawa M, Okamoto Y, Fukui H, Nakano H . Effect of dibutyryl cyclic AMP on the cyclin-dependent kinase inhibitor p27Kip1 in the human hepatoma cells PLC/PRF/5. Anticancer Res 1997; 17:3287–3291.
Miller MJ, Rioux L, Prendergast GV, Cannon S, White MA, Meinkoth JL . Differential effects of protein kinase A on Ras effector pathways. Mol Cell Biol 1998; 18:3718–3726.
Li H, Robinson PJ, Kawashima S, Funder JW, Liu JP . Differential regulation of MAP kinase activity by corticotropin-releasing hormone in normal and neoplastic corticotropes. Int J Biochem Cell Biol 1998; 30:1389–1401.
Dumaz N, Hayward R, Martin J, et al. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res 2006; 66:9483–9491.
Frodin M, Peraldi P, Van Obberghen E . Cyclic AMP activates the mitogen-activated protein kinase cascade in PC12 cells. J Biol Chem 1994; 269:6207–6214.
Bos JL . Epac: a new cAMP target and new avenues in cAMP research. Nature reviews. Mol Cell Biol 2003; 4:733–738.
Busca R, Abbe P, Mantoux F, et al. Ras mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J 2000; 19:2900–2910.
Kiermayer S, Biondi RM, Imig J, et al. Epac activation converts cAMP from a proliferative into a differentiation signal in PC12 cells. Mol Biol Cell 2005; 16:5639–5648.
Jivan A, Earnest S, Juang YC, Cobb MH . Radial spoke protein 3 is a mammalian protein kinase A-anchoring protein that binds ERK1/2. J Biol Chem 2009; 284:29437–29445.
Diviani D, Soderling J, Scott JD . AKAP-Lbc anchors protein kinase A and nucleates Galpha 12-selective Rho-mediated stress fiber formation. J Biol Chem 2001; 276:44247–44257.
Smith FD, Langeberg LK, Cellurale C, et al. AKAP-Lbc enhances cyclic AMP control of the ERK1/2 cascade. Nat Cell Biol 2010; 12:1242–1249.
Dumaz N, Marais R . Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J Biol Chem 2003; 278:29819–29823.
Wu J, Dent P, Jelinek T, Wolfman A, Weber MJ, Sturgill TW . Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3′,5′-monophosphate. Science 1993; 262:1065–1069.
Garnett MJ, Rana S, Paterson H, Barford D, Marais R . Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell 2005; 20:963–969.
Tzivion G, Luo Z, Avruch J . A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature 1998; 394:88–92.
Marquette A, André J, Bagot M, Bensussan A, Dumaz N . ERK and PDE4 cooperate to induce RAF isoform switching in melanoma. Nat Struct Mol Biol 2011; 18:584–591.
Balmanno K, Millar T, McMahon M, Cook SJ . DeltaRaf-1:ER* bypasses the cyclic AMP block of extracellular signal-regulated kinase 1 and 2 activation but not CDK2 activation or cell cycle reentry. Mol Cell Biol 2003; 23:9303–9317.
Pearson GW, Earnest S, Cobb MH . Cyclic AMP selectively uncouples mitogen-activated protein kinase cascades from activating signals. Mol Cell Biol 2006; 26:3039–3047.
Kraus AC, Ferber I, Bachmann SO, et al. In vitro chemo- and radio-resistance in small cell lung cancer correlates with cell adhesion and constitutive activation of AKT and MAP kinase pathways. Oncogene 2002; 21:8683–8695.
Sun S, Schiller JH, Spinola M, Minna JD . New molecularly targeted therapies for lung cancer. J Clin Invest 2007; 117:2740–2750.
Ravi RK, Weber E, McMahon M, et al. Activated Raf-1 causes growth arrest in human small cell lung cancer cells. J Clin Invest 1998; 101:153–159.
Ma PC, Tretiakova MS, Nallasura V, Jagadeeswaran R, Husain AN, Salgia R . Downstream signalling and specific inhibition of c-MET/HGF pathway in small cell lung cancer: implications for tumour invasion. Br J Cancer 2007; 97:368–377.
Arcaro A, Khanzada UK, Vanhaesebroeck B, Tetley TD, Waterfield MD, Seckl MJ . Two distinct phosphoinositide 3-kinases mediate polypeptide growth factor-stimulated PKB activation. EMBO J 2002; 21:5097–5108.
Bondzi C, Litz J, Dent P, Krystal GW . Src family kinase activity is required for Kit-mediated mitogen-activated protein (MAP) kinase activation, however loss of functional retinoblastoma protein makes MAP kinase activation unnecessary for growth of small cell lung cancer cells. Cell Growth Differ 2000; 11:305–314.
Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A . Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003; 4:181–189.
Sutherland KD, Proost N, Brouns I, Adriaensen D, Song TY, Berns A Berns A . Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011; 19: 754–764.
Blackhall FH, Pintilie M, Michael M, et al. Expression and prognostic significance of kit, protein kinase B, and mitogen-activated protein kinase in patients with small cell lung cancer. Clin Cancer Res 2003; 9:2241–2247.
Nayeem N, Kerr F, Naumann H, Linehan J, Lovestone S, Brandner S . Hyperphosphorylation of tau and neurofilaments and activation of CDK5 and ERK1/2 in PTEN-deficient cerebella. Mol Cell Neurosci 2007; 34:400–408.
Bost F, Aouadi M, Caron L, et al. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes 2005; 54:402–411.
Rodriguez A, Durán A, Selloum M, et al. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab 2006; 3:211–222.
Bamford S, Dawson E, Forbes S, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer 2004; 91:355–358.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is cited by
-
Genetic variants in Ras/Raf/MEK/ERK pathway are associated with gastric cancer risk in Chinese Han population
Archives of Toxicology (2020)
-
The lack of Raf-1 kinase feedback regulation enhances antiapoptosis in cancer cells
Oncogene (2017)
-
The nuclear translocation of ERK1/2 as an anticancer target
Nature Communications (2015)
-
C-Raf deficiency leads to hearing loss and increased noise susceptibility
Cellular and Molecular Life Sciences (2015)
-
Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling
Nature Cell Biology (2014)
