
Abstract
Both transcription and post-transcriptional processes, such as alternative splicing, play crucial roles in controlling developmental programs in metazoans. Recently emerged RNA-seq method has brought our understanding of eukaryotic transcriptomes to a new level, because it can resolve both gene expression level and alternative splicing events simultaneously. To gain a better understanding of cellular differentiation in gonads, we analyzed mRNA profiles from Drosophila testes and ovaries using RNA-seq. We identified a set of genes that have sex-specific isoforms in wild-type (WT) gonads, including several transcription factors. We found that differentiation of sperms from undifferentiated germ cells induced a dramatic downregulation of RNA splicing factors. Our data confirmed that RNA splicing events are significantly more frequent in the undifferentiated cell-enriched ba g of m arbles (bam) mutant testis, but downregulated upon differentiation in WT testis. Consistent with this, we showed that genes required for meiosis and terminal differentiation in WT testis were mainly regulated at the transcriptional level, but not by alternative splicing. Unexpectedly, we observed an increase in expression of all families of chromatin remodeling factors and histone modifying enzymes in the undifferentiated cell-enriched bam testis. More interestingly, chromatin regulators and histone modifying enzymes with opposite enzymatic activities are coenriched in undifferentiated cells in testis, suggesting that these cells may possess dynamic chromatin architecture. Finally, our data revealed many new features of the Drosophila gonadal transcriptomes, and will lead to a more comprehensive understanding of how differential gene expression and splicing regulate gametogenesis in Drosophila. Our data provided a foundation for the systematic study of gene expression and alternative splicing in many interesting areas of germ cell biology in Drosophila, such as the molecular basis for sexual dimorphism and the regulation of the proliferation vs terminal differentiation programs in germline stem cell lineages. The GEO accession number for the raw and analyzed RNA-seq data is GSE16960.
Similar content being viewed by others


Introduction
Drosophila melanogaster is an excellent model organism for studying the molecular mechanisms underlying cellular differentiation. Of all cell types, germ cells are unique in their ability to produce the next generation of an organism upon fertilization 1. The Drosophila male and female germlines are stereotypical adult stem cell lineages that provide powerful model systems for studying the molecular mechanisms that regulate stem cell maintenance vs differentiation 2, 3, 4, 5, 6, 7. Recent studies using loss-of-function assays for individual genes indicate that a unique chromatin structure in germline stem cells (GSCs) is critical for their self-renewal 8, 9. Previous microarray analysis also demonstrated that GSCs express specific isoforms of genes, which encode components distinct from those in canonical transcriptional or translational machinery 10. These data implicate alternative splicing in the regulation of GSC's maintenance.
Alternative splicing, through which a single pre-mRNA gives rise to different mature mRNAs, significantly increases protein diversity in higher eukaryotes 11, 12. Several well-known examples of how splicing regulates cellular differentiation during Drosophila development come from studying sexual development of somatic tissues, where the X chromosome to autosome ratio is fundamental in the establishment of somatic sexual identity through sex-specific gene splicing 13, 14, 15, 16, 17. Previous microarray studies of gene expression in whole flies reveal a broad spectrum of sex-specific expression of alternatively spliced isoforms 18. However, those studies cannot resolve the difference between somatic tissues and gonads. Recent exon-specific microarray studies have demonstrated a handful of genes that are sex-specifically spliced in Drosophila gonads, demonstrating that alternative splicing may also contribute to sex determination of gonads 19. On the other hand, the unequal dosage of X chromosomes between males and females necessitates compensatory mechanisms for X chromosomal gene expression. Through a process known as dosage compensation, the transcription level of genes on the single X chromosome in males is doubled to match the level of their expression in females 20. Previous microarray analysis of gene expression has shown that male-biased genes are depleted on the X chromosome 21, 22, 23. However, it is not clear whether such a preferential gene distribution applies to other groups of differentially expressed genes, such as stage-specific genes during gametogenesis.
Extensive global gene expression studies over the past decade, which have relied primarily on hybridization-based (e.g., microarray 24) and Sanger sequencing-based (e.g., serial analysis of gene expression (SAGE) 25) techniques, have described transcriptomes in a variety of cell types and developmental stages. However, these techniques are not optimal to resolve different isoforms of genes. Previous studies of alternative splicing mainly relied on expressed sequence tag (EST) sequencing data, which is very difficult to reach genome-wide coverage. Recently developed massively parallel signature DNA sequencing technologies have been applied to profile transcriptomes in yeast 26, 27, plant 28, and mammalian cells 29, 30, 31. The single-nucleotide resolution data from RNA-seq simultaneously provides information about mRNA levels and alternatively spliced isoforms (reviewed in Wang et al.32).
To obtain a comprehensive understanding of gene expression and splicing events during sexual and cellular differentiation of Drosophila gonads, we used the RNA-seq technique to profile mRNAs in wild-type (WT) gonads and mutant gonads enriched with undifferentiated cells, from both male and female adult flies. Our high-resolution, genome-wide transcriptome data revealed the existence of a large number of genes that exhibit differential splicing in male and female gonads, suggesting that alternative splicing may also be a critical mechanism underlying sexual dimorphism in gonads. A dramatic increase in the expression of all families of chromatin modifying enzymes, including both ATP-dependent chromatin remodeling factors (Brahma associated proteins (BAPs) and ATP-dependent nucleosome remodeling factor (NURF)) and histone modifying enzymes (histone methyltransferases (HMTs), histone demethylases (HDMTs), histone acetyltransferases (HATs), and histone deacetylases (HDACs)), was detected in the undifferentiated cell-enriched testis, indicating a need for the dynamic regulation of chromatin structure in those cells. At the same time, splicing factors were expressed at high levels in the undifferentiated cell-enriched testis, suggesting that alternative splicing is important in maintaining the undifferentiated status of those cells in testis. In contrast, genes required for meiosis and terminal differentiation were mainly regulated at the transcriptional level, and not by alternative splicing, in the differentiated cells of testis. Interestingly, we found that the differentiation genes in testis have a significantly biased distribution on the 2L autosomal arm. In contrast, these genes are the most underrepresented on the X chromosome, indicating a potential mechanism for them to escape X-inactivation, which is thought to occur during late spermatocyte stage. In summary, our high-resolution and genome-wide RNA-seq data provided a platform to study sexual and cellular differentiation of Drosophila gonads at a molecular level.
Results
Generation of sex- and stage-specific gonadal transcriptomes using RNA-seq
To characterize the molecular signatures of sexual and cellular differentiation during gametogenesis, we isolated female and male gonadal poly (A) RNAs from four different adult flies (Figure 1A): ovaries and testes from the bam mutant flies and ovaries and testes from WT flies. In bam mutant gonads, the transition from undifferentiated cells to differentiating cells is abolished and the tissues are enriched with over-proliferative undifferentiated cells, including GSCs, transit-amplifying spermatogonial cells, and somatic cells 33, 34. Although the bam mutant gonads have some abnormal features, they are highly enriched with early-staged germ cells, and thus, have been used in previous microarray analysis to study molecular characteristics of undifferentiated cells in gonads 10, 35.

Generation of gonadal transcriptomes using RNA-seq. (A) The four samples used for RNA-seq and four pair-wise comparisons in two categories to investigate: (1) sex-biased, (2) stage-specific patterns of gene expression and alternative splicing. (B) The anti-RNA Pol II ChIP-seq results using the bam testis sample. The five gene groups were classified according to their RPKM value based on RNA-seq results. The thresholds for low, moderate, and high expression level were set up to reach the same number of genes in each group.
Using these gonadal samples, we performed RNA-seq analysis using the Illumina/Solexa genome analyzer, as described in the Materials and Methods and Supplementary information, Figure S1A. We retrieved about 10 million non-redundant unique reads per sample to reach a sequencing depth sufficient to cover the entire predicted fly transcriptome ∼10 times (Table 1). We normalized transcript abundance for all expressed genes as sequencing reads/per kilobase merged exonic region/per million mapped reads (RPKM value 29, see Supplementary information, Figure S1B and Materials and Methods). To evaluate how well the RPKM value reflects transcriptional activity of genes, we used ChIP-seq (chromatin immunoprecipitation followed by high-throughput sequencing) to plot the enrichment of RNA Pol II at the transcription start sites (TSSs) for five groups of genes in the bam testis sample, classified according to their RPKM values 30 (Figure 1B). The Pol II occupancy at TSSs positively correlated with genes' RPKM values. The silent group (RPKM = 0) showed no Pol II enrichment.
Using the RPKM values of individual genes (Supplementary information, Table S1), we performed four pair-wise comparisons between different samples in two categories (Figure 1A): (1) sex-biased genes in bam-mutant and WT gonads to examine key regulators for sexual dimorphism in gonads; (2) genes that are expressed in a stage-specific manner in male and female gonads, to study stem cell or undifferentiated cell-enriched factors, as well as genes required for cellular differentiation of gametes.
The WT gonads demonstrated an outstanding sex-biased gene expression pattern, shown as a low correlation coefficient (r = 0.182 for WT ovary vs WT testis comparison). The sexual difference in undifferentiated cell-enriched bam gonads was much smaller (r = 0.716 for bam ovary vs bam testis comparison). In addition, more differential gene expression was observed in testes than in ovaries when comparing gonads with different-staged germline and somatic cells: testis samples showed much higher variation (r = 0.242 for bam vs WT testis) than ovary samples (r = 0.865 for bam vs WT ovary). A possible explanation for such a difference is that we used flies that were within 1 day post-eclosion, which is sufficient for testis to reach full-term spermatogenesis, but may be too early for ovary to have mature eggs 36, 37.
To further evaluate the data quality, we compared our RNA-seq results with the existing gene expression profiling data using microarray. We found that our data were largely consistent with the microarray data 22 (http://www.flyatlas.org/). For example, in the WT testis vs WT ovary comparison (Supplementary information, Figure S2), greater than 77% (in most cases, greater than 89%) of the sex-biased genes identified by microarrays overlapped with the same category of genes identified by RNA-seq, indicating that the RNA-seq method largely recapitulated the microarray results and will be useful to identify other differentially expressed genes in gonads. Our data also showed that ∼56% of all genes in WT gonads and 21% in bam-mutant gonads showed sex-biased expression patterns (≥ 2-fold difference between sexes). These data were consistent with the previous work that indicates that approximately half of the Drosophila genes have sex-biased expression pattern 18 and WT gonads contain most of them 38, 39, 40.
Enrichment of chromatin regulators in undifferentiated cell-enriched bam testis
The differentiation of germ cells, especially in testis, is characterized by a dramatic morphological change and genome-wide chromatin condensation with replacement of histones by protamines 41. To probe the molecular circuitries regulating gametes differentiation programs, gene expression profiles between WT and bam mutant gonads were compared in both males and females.
These comparisons indicated that spermatogenesis has a distinct gene expression profile switch from undifferentiated cell-enriched bam testis to fully differentiated WT testis: We found that 501 and 1 894 genes were uniquely expressed in bam and WT testis, respectively (Figure 2A). These results were consistent with the thought that a large pool of meiotic and terminal differentiation genes is transcribed in spermatocytes (Fuller 42 and Fuller, MT and White-Cooper, H personal communications). Examples of such genes included the testis-specific TAF homologs (tTAFs) and components of a testis-specific version of the MIP/DREAM complex (tMAC), which are remarkably upregulated in WT testis (Figure 2B) 42, 43, 44, 45.

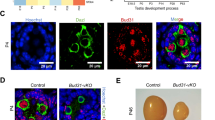
Enrichment of the chromatin regulators in undifferentiated cell-enriched bam testis. (A) Heat map showing genes that are uniquely expressed in both bam testis vs WT testis, and bam ovary vs WT ovary comparisons. Unique expression means that a certain gene is expressed in one sample (RPKM ≥ 1), but silent in another sample (RPKM < 0.5). The RPKM cutoffs for expressed and silent genes are based on Gan et al. 98. Heat scale: log2RPKM. To calculate the log2RPKM values of individual gene, all their original RPKM values were added by a pseudo-count of 1. (B) Enriched chromatin remodeling/modifying factors and histone modifying enzymes in bam testis. Abbreviations: PcG, polycomb group complex; PRC1 and PRC2, polycomb repressive complex 1 and 2; TrxG, trithorax group complex; HMT, histone methyltransferase; HAT, histone acetyltransferase; HDAC, histone deacetylase; tTAFs, testis TAF homologs; tMAC, a testis-specific version of the MIP/DREAM complex; meiotic genes and TD, terminal differentiation genes for spermiogenesis. (C) In situ data using antisense riboprobes that recognize the trx (HMT) gene and lid (HDMT) gene in WT testis. There is a coenrichment of the trx and lid transcripts in undifferentiated cells located at the tip of the WT testis, indicated by the black arrows. (D) In situ data using antisense riboprobes that recognize the Pcaf (HAT) gene and Rpd3 (HDAC) gene in WT testis. There is coenrichment of the Pcaf and Rpd3 transcripts in undifferentiated cells located at the tip of the WT testis, indicated by the black arrows.
Previous studies have suggested that the undifferentiated status of female GSCs is maintained by a unique chromatin structure 8, 9, 46. To examine the potential epigenetic mechanisms that maintain the undifferentiated status of male GSCs and spermatogonia, we compared the expression levels of all families of chromatin remodeling factors and histone modifying enzymes in bam testis vs WT testis. We found that 62.5% (20 out of 32) of chromatin remodeling factors and 70.2% (40 out of 57) of histone modifying enzymes are highly enriched in the undifferentiated cells of bam testis (Table 2). E.g., BAP60 and BAP55, components of the ATP-dependent SWI/SNF family of chromatin remodeling BAP complexes 47, 48, were elevated 6.1-fold and 5.8-fold, respectively, in bam testis compared to WT testis. Nurf-38, a subunit of the NURF complexes 49, 50, was elevated 3.4-fold in bam testis compared to WT testis (Table 2). The Polycomb group (PcG) and Trithorax group (TrxG) complexes, as well as other histone modifying enzymes, including HMTs, HDMTs, HATs, and HDACs, were also remarkably elevated in undifferentiated cells in bam testis. Interestingly, chromatin modifying enzymes that have antagonizing functions were coenriched in bam testis. For example, the activities of the PcG and TrxG protein complexes counteract with each other in the determination of cell fate 51, 52. We detected a 4.1-fold enrichment of a key PcG component Enhancer of zeste (E(z)) and a 3.3-fold enrichment of a critical TrxG component trithorax (trx) in bam testis, compared to WT testis (Figure 2B and Table 2). In addition, genes encoding histone modifying enzymes that have opposite biochemical activities, such as the H3K4me3 HMT (e.g., trx) 53 and HDMT (e.g., little imaginal disc or lid: 5.6-fold enrichment) 54, 55, 56, as well as HATs (e.g., Pcaf: 4.9-fold enrichment) 57 and HDACs (e.g., Rpd3: 15.4-fold enrichment) 58, were both highly expressed in bam testis compared to WT testis. Interestingly, the lid gene was genetically identified as a TrxG gene 59, indicating that it may act with trx in a coordinated manner to antagonize PcG activities and regulate male germ cell differentiation. Recent studies have also demonstrated that the HAT and HDAC enzymes act together to regulate gene transcription 60, and different histone enzymes associate with each other and affect their respective enzymatic activities 61. Consistent with those findings, here our data demonstrated that chromatin modifying enzymes with opposite activities were coenriched and may act cooperatively to regulate the undifferentiated status of male germ cells.
To further validate these RNA-seq results, we performed in situ hybridization using gene-specific antisense riboprobes for a number of chromatin regulators. Indeed, we found coenrichment of the trx and lid transcripts in undifferentiated cells located at the tip of the WT testis (Figure 2C). The same coenrichment pattern was also obtained for the antagonizing Pcaf and Rpd3 genes (Figure 2D). These data confirmed the RNA-seq results and implicated that the chromatin status of undifferentiated cells in testis may be dynamically regulated by balanced activities of counteracting chromatin regulators and histone modifying enzymes. In contrast, among the 925 genes that are uniquely expressed in bam ovary (Figure 2A), only two chromatin regulator genes (tna and Iswi) were enriched in the bam ovary compared to WT ovary. These observations were consistent with previous reports that female GSCs have a relatively repressed transcriptome 10, 62 and possibly a repressive chromatin landscape.
In summary, our data suggest that the status of undifferentiated cells in bam testis is associated with high expression level of a cohort of chromatin regulators, which may be critical for the maintenance of the unique molecular identities and cellular behavior of undifferentiated male GSCs and spermatogonial cells, which have been shown to retain the plasticity to dedifferentiate to become GSC-like cells 7, 63, 64.
RNA splicing factors are highly enriched in undifferentiated cell-enriched bam testis
Interestingly, we found significant enrichment of splicing factors in the bam testis: ∼56.9% of genes that encode characterized or putative splicing factors were enriched at least 2-fold in bam testis relative to WT testis (177 out of 311, Figure 3A and Supplementary information, Table S2), which was significantly higher than random distribution (P < 10−15). The bam testis-enriched splicing factors included seven genes encoding SR proteins, which regulate splice site selection (Supplementary information, Table S2). In contrast, only 8.4% of splicing factors were enriched in WT testis compared to bam testis (26 out of 311, Figure 3A and Supplementary information, Table S2), which was significantly lower than random distribution (P < 10−8). These data suggested that the undifferentiated cells in testis may be associated with a genome-wide increase of splicing activities.

Enhanced splicing activities in undifferentiated cell-enriched bam testis. (A) Expression of splicing factors is upregulated in bam testis compared to WT testis. The red and green lines are the 2-fold cutoff lines. 177 splicing factors are 2-fold more enriched in bam testis relative to WT testis, while 26 splicing factors are 2-fold more enriched in WT testis relative to bam testis. The enrichment of splicing factors in bam testis is significant (P < 10−15). P-value was calculated using the chi-square test. (B) Enhanced overall splicing activities in bam testis compared to WT testis, shown by the CDF plot. P-value was calculated using one-sided Kolmogorov-Smirnov test. (C) Percentage of differentiation genes as single-isoform vs multi-isoform genes (Differentiation genes are defined as silent in bam testis (RPKM in bam testis < 0.5), but are expressed in WT testis (RPKM in WT testis ≥ 1)). Only 13% of the differentiation genes are multi-isoform genes, which is significantly (P < 10−15) lower than the 23% genome-wide proportion of multi-isoform genes, based on the Ensembl annotation. The P-value was calculated using one-sided Fisher exact test.
Enhanced alternative splicing activities in undifferentiated cell-enriched bam testis
To test the above hypothesis, we used gene entropy (S) as a measure of overall splicing complexity at any given splicing locus (Materials and Methods and Ritchie et al.65). We computed gene entropy using the formula S = −∑pk log2 pk. The pk is the frequency of a particular transcribed isoform k, which is estimated from the RNA-seq data (Materials and Methods and Supplementary information, Table S3). We then used normalized genes' entropies to plot a “Cumulative Distribution Function (CDF)” for each sample (Materials and Methods).
We first examined alternative splicing activities in bam gonads and compared the results with those in WT gonads. Indeed, we found that there were 23% more alternatively spliced genes in bam testis compared to WT testis (Table 1). Consistently, the CDF plot of bam testis shifted to the right side of WT testis, indicating an overall higher complexity of transcripts in bam testis (P < 0.005, Figure 3B). Similarly, but less significantly, results were obtained by comparing the bam ovary vs WT ovary (P < 0.02, data not shown).
The significantly decreased expression of splicing factors in WT testis (Figure 3A) implicated that single-transcript genes may be popular for terminal differentiation genes in testis (RPKM < 0.5 in bam testis and RPKM ≥ 1 in WT testis). Indeed, we found that the percentage of multi-isoform genes is significantly (P < 10−15) underrepresented among terminal differentiation genes in testis (Figure 3C): only 13% of testis differentiation genes are multi-isoform genes, whereas 23% of all Ensembl annotated genes are multi-isoform genes. In addition, when we computed the ratio (percentage) of the intronic region size relative to the total gene size, we found a unique feature for testis differentiation genes: the mean of the intron size to the entire gene size ratio for all Drosophila genes is ∼25%. However, for the terminal differentiation genes in testis, this ratio was significantly reduced to 19.3% (P < 10−16). These intriguing results indicated that this particular group of genes might have evolved to become the simplest single-isoform ones, which could be the molecular mechanism that ensures efficient and coordinated transcription of a cohort of genes required for meiosis and spermiogenesis. However, this finding appeared to be in contrast with previous studies in mammals, which have suggested that alternative splicing is prevalent in testis 66, 67. This apparent discrepancy could be due to mixed-stage germ cells in the mammalian studies, incomplete coverage of the mammalian EST libraries, or a difference between mammals and flies during evolution.
Sex-specific isoforms in bam and WT gonads
Alternative splicing is well known to play a critical role in sexual differentiation in Drosophila somatic tissues 17, 68. However, sex determination of germ cells in gonads is not as well understood, but is thought to take a different route and rely on signals emanating from surrounding somatic cells 69, 70. Interestingly, recent studies have demonstrated that alternative splicing may also contribute to sexual differentiation of gonads, with a set of representative examples 19. Here, we used the RNA-seq data to identify sex-specific isoforms genome widely in gonads. We analyzed uniquely detectable isoforms of each alternatively spliced gene in all four samples (Materials and Methods and Supplementary information, Table S3). We then plotted each alternatively spliced gene for the presence or absence of sex-specific isoforms in both bam testis vs bam ovary, and WT testis vs WT ovary comparisons, respectively. Our analysis revealed 462 genes with sex-specific isoforms in bam testis vs bam ovary comparison (Figure 4A and Supplementary information, Table S4-1). We also identified 614 genes that have sex-specific isoforms in WT testis and WT ovary (Figure 4B and Supplementary information, Table S4-2). Surprisingly, p53 was among these genes with different isoforms in WT male and female gonads (Table 3 and Supplementary information, Table S4-2). Using real-time PCR assays with isoform-specific primers, we found that the p53-RA isoform was 2-fold more enriched in WT ovary than in WT testis; whereas the p53-RB isoform was 14.6-fold more enriched in WT testis than in WT ovary (Figure 4C and 4D). The p53 gene is well known for its regulation of cell cycle and programmed cell death in response to cellular stresses 71, 72. The Drosophila p53 was also known to regulate primordial germ cell development in embryos 73. Our discovery that there are distinct p53 isoforms in WT testis and ovary implicated potentially dimorphic roles of p53 in regulating apoptosis, which could contribute to distinct cellular differentiation pathways of male and female gametes.

Visualization of sex-specific isoforms of individual genes in bam and WT testis vs ovary comparisons. (A) Genes with sex-specific isoforms in bam testis vs bam ovary. All genes with ≥ 2 uniquely detectable isoforms (Materials and Methods) were analyzed for the presence (Red, R) and absence (Blue, B) of sex-specific isoforms. Presence (red) in “both” category indicated that the corresponding gene has at least one isoform expressed in both bam testis and bam ovary samples. The gene numbers of each class were labeled on the y axis of the panel. The RRR, RBR, BRR, and RRB classes had at least one sex-specific isoform, therefore were labeled as black numbers (see Supplementary information, Table S4-1 for details.). (B) Genes with sex-specific isoforms in WT testis vs WT ovary. The presence (R) and absence (B) of sex-specific isoforms for each individual gene were analyzed in WT testis and WT ovary samples; the presence (red) in both somatic samples indicated a lack of sex specificity for a particular isoform(s) of an individual gene (Supplementary information, Table S4-2). (C) The WT ovary-biased p53 isoform is CG33336-RA or FBtr0084359. The WT testis-biased p53 isoform is CG33336-RB or FBtr0084360. (D) Real-time RT-PCR using isoform-specific primer sets showed that p53-RA is about 2-fold more enriched in WT ovary compared to WT testis, while p53-RB is ∼14.6-fold more enriched in WT testis compared to WT ovary. The level of both p53-RA and p53-RB were normalized to the total p53 level, using a primer set that amplifies a common region of both isoforms (Materials and Methods). The total p53 level in WT ovary and WT testis was also shown, which has been normalized to the sample with a smaller RPKM value (WT testis in this case, Supplementary information, Table S1). (E) The WT ovary-biased Rab14 isoform is CG4212-RA or FBtr0080626. The WT testis-biased Rab14 isoform is CG4212-RB or FBtr0080627. (F) Real-time RT-PCR using isoform-specific primer sets showed that Rab14-RA is about 13.9-fold more enriched in WT ovary compared to WT testis, while Rab14-RB is ∼171.4-fold more enriched in WT testis compared to WT ovary. The levels of both Rab14-RA and Rab14-RB were normalized to the total Rab14 level, using a primer set that amplifies a common region of both isoforms (Materials and Methods). The total Rab14 level in WT ovary and WT testis was also shown, which has been normalized to the sample with a smaller RPKM value (WT ovary in this case, Supplementary information, Table S1).
Another interesting example is the Rab14 gene (Table 3 and Supplementary information, Table S4-2). Our real-time PCR analyses revealed that the two Rab14 isoforms demonstrated the opposite enrichment in WT ovary (Rab14-RA, with 13.9-fold enrichment in ovary) and WT testis (Rab14-RB, with 171.4-fold enrichment in testis, Figure 4E and 4F). The Rab genes encode small guanosine triphosphatases (GTPases), which have important roles in regulating vesicle trafficking and actin filament assembly in flies 74, 75. Interestingly, certain Rab gene, such as the Rab11, has been shown to maintain the identities of female GSCs 76. It will be intriguing to explore whether Rab14 may regulate germ cell dimorphism or have dimorphic functions through its distinct isoforms. More examples of genes that exhibit sex-specific (biased) isoforms are shown in Supplementary information, Figure S3. In summary, our data indicated that sex-specific isoforms may contribute to sex-specific gametogenesis and provide a splicing atlas for further studies.
Splicing factors (or putative splicing factors) themselves exhibit sex- and stage-specific isoforms in gonads
It has been reported that splicing factors themselves are regulated by alternative splicing during C. elegans development 77. To obtain a comprehensive understanding of this phenomenon in Drosophila gonads, we searched all 311 genes that encode characterized or putative splicing factors (Supplementary information, Table S2) for distinct isoforms in both bam and WT gonads. Indeed, we found that 21 of them have differential isoforms in at least one of the four pair-wise comparisons (Supplementary information, Table S4-1 to Table S4-4). One interesting example is the exuperantia (exu) gene (Table 3), which regulates RNA localization 78, 79 and possibly splicing as well (Supplementary information, Table S2). The exu gene exhibits germline-specific expression pattern in adult flies 80 and has been demonstrated to have male germ cell-specific splicing under the control of the Transformer 2 (Tra2) splicing factor 81. Our RNA-seq data confirmed previous reports that the exu-RC is a testis-specific isoform. In addition, using real-time PCR assays, we found that the exu-RA is a testis-biased isoform (with 4.9-fold enrichment in WT testis) and the exu-RB is an ovary-biased isoform (with 94.3-fold enrichment in WT ovary, Figure 5A and 5B). The differences of all exu transcripts are located at the untranslated regions (UTRs), which may lead to different Exu protein levels in male and female germ cells. It will be interesting to examine whether such a difference may allow Exu to execute sex-specific functions, such as splicing, in germ cells.

Examples of characterized or putative splicing factors that have sex- or stage-specific isoforms. (A) UCSC snapshots show that the exu gene has sex-specific and sex-biased isoforms: exu-RC is WT testis-specific, exu-RA is WT testis-biased, and exu-RB is WT ovary-biased isoforms. (B) Real-time RT-PCR results showed that exu-RA is 4.9-fold more enriched in WT testis compared to WT ovary, while exu-RB is ∼94.3-fold more enriched in WT ovary compared to WT testis. The levels of both exu-RA and exu-RB were normalized to the total exu level, using a primer set that amplifies a common region of all three isoforms (Materials and Methods). The total exu level in WT ovary and WT testis was also shown, which has been normalized to the sample with a smaller RPKM value (WT ovary in this case, Supplementary information, Table S1). (C) UCSC snapshots show that the imp gene is expressed in both undifferentiated spermatogonia-enriched bam testis and differentiating spermatids-containing WT testis. The imp gene has stage-specific isoforms: imp-RA/RB/RC is bam testis-specific and imp-RG/RH is WT testis-specific. (D) Real-time RT-PCR results showed that imp-RA/RB/RC is 40.2-fold more enriched in bam testis compared to WT testis, while imp-RG/RH is ∼1 966-fold more enriched in WT testis compared to bam testis. The level of both subsets of imp isoforms were normalized to the total imp level using a primer set that amplifies a common region of all isoforms (Materials and Methods). The total imp level in bam testis and WT testis was also shown, which has been normalized to the sample with a smaller RPKM value (bam testis in this case, Supplementary information, Table S1).
Another interesting example showed that a splicing factor had stage-specific isoforms (Supplementary information, Table S4-3 and Table S5). The IGF-II mRNA-binding protein (Imp) is a component of the Drosophila spliceosomal complex 82. The Imp gene is expressed in both undifferentiated spermatogonia and differentiating spermatids 83. Our RNA-seq data and real-time PCR validation experiments revealed both bam testis-specific isoforms (imp-RA, imp-RB, and/or imp-RC with 40.2-fold enrichment in bam testis) and WT testis-specific isoforms (imp-RG and/or imp-RH with 1 966-fold enrichment in WT testis, Figure 5C and 5D), indicating that it may regulate splicing in both staged cells using distinct isoforms. Overall, our results suggested that a subset of splicing factors (or putative splicing factors) may regulate cell type-specific splicing through their own sex- or stage-specific isoforms.
Biased distribution of testis terminal differentiation genes on the 2L chromosomal arm
We next investigated whether and how chromosomal territories affect gene expression by studying chromosomal distribution of stage-specific genes during spermatogenesis. In mammals, it has been reported that the spermatogonial genes are overrepresented on the X chromosome 84, 85, 86. Here, we mapped spermatogonia-enriched genes based on the comparison of bam testis vs WT testis (Figure 6A). We found that the bam testis (thus spermatogonia)-enriched genes were significantly enriched on the X chromosome (P < 10−15), consistent with what has been reported from mammalian studies. Interestingly, this biased gene distribution on the X chromosome was reversed on the 2L arm, where the spermatogonia-enriched genes were the least frequent (P < 10−7). In contrast, terminal differentiation genes that are highly expressed in WT testis were significantly underrepresented on the X chromosome (P < 10−5), and were found mostly enriched on the 2L arm (P < 10−5) (Figure 6B). X-inactivation is thought to occur during the late spermatocyte stage 87, which could have provided the selective pressure for differentiation genes to translocate from the X chromosome to autosomes, where they could avoid inactivation. These data suggested that in addition to the X chromosome, the 2L arm is critical in the determination of proper gene expression during spermatogenesis. And the fact that the X chromosome and 2L chromosomal arm were reciprocally favored or avoided suggested that the gene distribution on these two chromosomes may have been subjected to selective pressures during evolution.

Preferential gene distribution of testis terminal differentiation genes on the 2L chromosomal arm. Preferential chromosomal distribution of stage-biased genes in testis on the X chromosome and the 2L chromosomal arm: percentage was calculated as the differentially expressed genes (≥ 2-fold change)/total genes on a particular chromosome arm (X, 2L, 2R, 3L, and 3R). To calculate the ratio of RPKM of bam testis/WT testis or WT testis/bam testis, the RPKM value was set to 0.5 if less than 0.5. P-value was calculated with the Pearson's chi-square test (Materials and Methods).
New features of the Drosophila gonadal transcriptomes
Our RNA-seq data revealed many new features of the Drosophila gonadal transcriptomes, including thousands of transcribed regions that are not included in current Flybase. We called them complementary-to-Flybase transcribed regions or CFTRs. In each sample, we identified ∼1 500-2 500 CFTRs (Materials and Methods). The average size of the CFTRs was ∼400 bp (Table 1). Most of the CFTRs were located in the intergenic regions (Figure 7A). On average, the expression level of CFTRs was ∼30-43% of that of the annotated genes (Figure 7B). To test whether the CFTRs have been predicted or identified, we compared them to the N-SCAN, CONTRAST, mRNA, and EST databases. We found that about 47-83% of the identified CFTRs in each sample were supported by the mRNA or EST databases, and about 15-20% of the identified CFTRs in each sample were present in predicted gene datasets (Supplementary information, Table S6 and Materials and Methods). Using an extensive BLAST search, we found that only 8.9–15.4% of the CFTRs contain known protein-coding sequences (Table 1, Supplementary information, Table S6 and Materials and Methods). The remaining ∼85% of the CFTRs could encode novel peptides or non-coding RNAs. Interestingly, analysis of the tissue specificity of the CFTRs revealed hundreds of specific sequences in each pair-wise comparison (Supplementary information, Figure S4). This result indicated that CFTRs are another source of differentially expressed transcripts in gonads.

New features of the Drosophila gonadal transcriptome. (A) Distribution of identified CFTRs at the intronic and intergenic regions, based on the Ensembl annotation (http://www.ensembl.org/index.html) and the most recently updated Flybase version (r5.19). *, Retained intronic CFTR must have a RPKM value greater than 10% of the RPKM(s) of the neighboring gene(s), to avoid contamination from pre-mRNAs. (B) Box plot of the expression level (log2RPKM) of annotated expressed genes and CFTRs in all four samples.
In summary, our data revealed many new features of the Drosophila gonadal transcriptomes, which will lead to a more comprehensive understanding of how differential gene expression and splicing regulate sexual and cellular differentiation of the fly gonads 88, 89, 90.
Discussion
Epigenetic regulation in male gonads
Emerging evidence indicates that embryonic stem cells maintain their identities by a unique transcription network and chromatin structure (reviewed in Boyer et al. 91 and Jaenisch and Young 92). However, it is not well understood whether adult stem cells, such as GSCs, maintain their unique features using a particular chromatin structure, and if so, how developmental programs change such a structure and regulate terminal differentiation (reviewed in Chen X 93 and Buszczak and Spradling 94). Previous studies in male germline lineage revealed that the cell type-specific tTAFs counteract Polycomb functions upon differentiation 44, which indicate that the chromatin structure in germ cells may switch upon the transition from undifferentiated to differentiating status. Here, our RNA-seq data demonstrated a remarkable enrichment of nearly all families of chromatin modifying enzymes, including both ATP-dependent chromatin remodeling factors and histone modifying enzymes in the undifferentiated cell-enriched testis, compared with fully differentiated testis. These data demonstrated that a dynamically regulated chromatin structure may be required for maintaining the undifferentiated status of male GSCs and transit-amplifying spermatogonial cells. In addition, developmentally programmed mechanisms are required for switching such a chromatin landscape in order to initiate the terminal differentiation gene expression program.
Enriched splicing factors in undifferentiated cell-enriched testis may contribute to stage-specific splicing events in testis
Differential gene expression and alternative splicing contribute significantly to pleiotropic, phenotypic, and behavioral features 10, 22, 35. In addition to the dynamic chromatin structure, our studies revealed enrichment of the majority of the splicing factors in the undifferentiated cell-enriched testis. The significantly increased splicing activities may collaborate with the highly expressed chromatin remodeling factors and histone modifying enzymes to maintain an intricate but sensitive transcription network through post-transcriptional mechanisms. Upon differentiation, the developmental program switches the gene network from undifferentiated to differentiating status in a unidirectional mode. The terminal differentiation genes that are turned on in differentiating cells are less dependent on splicing factors. The lower transcript complexity of this group of genes may contribute to the higher degree of transcriptional efficiency, to coordinate the coexpression of a large number of genes in spermatocytes 42. It is not clear how these stage-specific features of transcriptomes are regulated. However, these phenomena are amenable to further genetic and molecular studies.
Sex-specific isoforms in gonads
Among all the phenotypic traits of Drosophila, sexual dimorphism is undoubtedly one of the most distinct features, and gonads are the most sexually distinguishable tissues. Despite a wealth of knowledge about sexual differentiation of soma, sexual differentiation of germ cells is not well understood. Our data demonstrate that WT gonads are not only the major tissues with sex-specifically expressed genes but also the tissues with prevalent sex-specific isoforms of genes (Figure 4B). Studying these genes, especially those with regulatory roles such as transcription factors or splicing factors, will undoubtedly shed light on how sexual dimorphism is established and maintained during gametogenesis. And our work has provided much needed information to address these intriguing biological questions at a molecular level in gonads.
In summary, we have discovered that the dynamic regulation of chromatin remodeling factors and histone modifying enzymes is characteristic of undifferentiated cell-enriched Drosophila testis. The sex-specific isoforms of critical transcription factors and splicing factors may contribute to the sexual differentiation of gonads. Our single base-pair resolution, genome-wide RNA-seq data provide a foundation for the systematic study of many interesting areas in Drosophila reproductive biology and stem cell biology, such as the molecular basis for sexual dimorphism of gonads and the regulation of proliferation vs terminal differentiation programs in GSC lineages.
Materials and Methods
Fly strains and tissue preparation
All fly stocks were grown in a 25 °C incubator with standard medium. The bam[1]/TM3 stock were obtained from Bloomington Drosophila Stock Centers. The bam[Δ86]/TM3 strain was a gift from Dr Allan C Spradling. The bam[114-97]/TM6B strain was a gift from Dr Margaret Fuller. Testes from bam[1]/bam[114-97]-mutant males and ovaries from bam[1]/bam[Δ86] females were dissected in DEPC-treated 1× PBS buffer in 20 min interval, followed by immediate snap freezing in liquid nitrogen. The WT testes and ovaries were dissected from y, w flies using the same method. All flies were less than 1 day post-eclosion. Noticeably, we found some accessory gland genes (e.g., Acp genes) in our bam testis RNA-seq dataset (Supplementary information, Table S1), which may be caused by technical difficulties to completely isolate testes from accessory glands from bam-mutant males. In fact, we found the same issue with previous microarray data using a similar fly strain 35, shown as high deviation among different biological replicates.
Library preparation for RNA-seq
Total RNA was extracted using TRIzol (Invitrogen, cat. no. 15596-018) following the manufacturer's instructions. Samples used for total RNA extraction were ∼200 pairs of bam testes (8.5 μg), ∼200 pairs of bam ovaries (6.9 μg), ∼200 pairs of WT testes (19 μg), and 45 pairs of WT ovaries (15 μg). DNA was degraded using 2 U of DNase I (Fermentas, cat. no. EN0521) at 37 °C for 20 min. The integrity of RNA was checked by gel electrophoresis (1% agarose).
From ∼10 μg total RNA for each sample, we performed two rounds of mRNA isolation using Dynabeads mRNA purification kit (Invitrogen, cat. no. 610-06), according to the manufacturer's instructions. The final mRNAs were eluted in 13.5 μl of 10 mM Tris-HCl (pH 7.5) and immediately used to generate the first strand cDNA, using 4 μl of random hexamers (ABI, cat. no. N8080127) and SuperScript II Reverse Transcription Kit (Invitrogen, cat. no. 18064-014) in a 30 μl final volume, following the manufacturer's instructions. The second strand cDNA was generated using the following recipe: 10 μl of 5× second strand buffer (500 mM Tris-HCl (pH 7.8), 50 mM MgCl2, 10 mM DTT), 30 nmol dNTPs (Invitrogen, cat. no. 18427-013), 2 U of RNase H (Invitrogen, cat. no. 18021-014) and 50 U of DNA Pol I (Invitrogen, cat. no. 18010-025). The entire reaction mixture was incubated at 16 °C for 2.5 h. The double-stranded DNA (dsDNA) was purified with QIAquick PCR purification kit (Qiagen, cat. no. 28106) and the concentration was quantified by a Qubit fluorometer (Invitrogen).
To generate libraries for sequencing, ∼300 ng dsDNA of each sample was fragmented by sonication using Bioruptor (Diagenode, UCD-200-TM-EX) under the following conditions: medium power output for 30 min in ice water. The resulting DNA fragments were analyzed by agarose gel to verify a ∼100-300 bp size range. Sequencing libraries were prepared as the follows: end-repair (DNA end-repair kit from Epicenter, cat. no. ER0720); A-tailing (300 ng dsDNA, 5 μl Thermo buffer, 10 nmol dATP, 15 U of Taq polymerase, at 70 °C for 30 min); Solexa adaptor ligation (300 ng dsDNA, 4 μl DNA ligase buffer, 1 μl Solexa adaptor mixture, 3 μl DNA ligase, at 70 °C overnight.); PCR (98 °C 10 s, 65 °C 30 s, 72 °C 30 s for 16 cycles; then additional 72 °C for 5 min) amplification with adaptor primers; and size selection (200-400 bp). Then the library dsDNA for each sample was used on Solexa 1G sequencer at a concentration of 10 ng per lane.
Primary annotation information
Drosophila exon annotation information was downloaded using BioMart from the Drosophila BDGP5.4, Ensembl database (release 50).
Preliminary analysis of short read data Short reads alignment and filtering
The quality-filtered 30 bp short sequence reads were aligned to the reference sequence consisting of dm3 Drosophila melanogaster genome plus a library of synthetic exon junction sequences using ELAND (Efficient Local Alignment of Nucleotide Data) software, allowing up to two mismatches with the reference sequence (Supplementary information, Figure S1B). The library of exon junction sequences was created as follows. Drosophila exon sequences were retrieved from Ensembl database (release 50). All possible pairs of exons that belonged to the same transcript were joined such that the genomic order of exons is aligned. A junction sequence consisted of the last 26 bp of 5′ exon and the first 26 bp of 3′ exon. Redundant sequences were removed from the resulting set of 52-bp exon junction sequences. The numbers of reads that aligned uniquely to the genome and exon junctions are shown in Table 1. To remove possible PCR amplification artifacts and to reduce confounding effects of systematically bad sequencing cycles in short sequence reads, we retained a single copy of each unique read. This filtering procedure yields the set of non-redundant unique reads.
Sequencing depths in different samples
There was some variability in sequencing depths in different tissue samples. At least two technical replicates were used and 10-13 million 30 bp sequencing reads were obtained per sample (Table 1). To characterize splicing and gene expression differences between samples in an unbiased way, we used the following procedure to equate the sequencing depths of different samples. We randomly sub-sampled ∼10 million non-redundant unique reads (300 million bp) in each of the four samples and used these reads for all downstream analyses, which was sufficient to cover the entire predicted fly transcriptome 10 times (∼30 million bp transcribed sequences according to Ensembl database release 50).
Calculation of gene expression level
For each tissue sample, we used the following procedure to compute a single number that summarizes overall expression level of a gene. All exon regions belonging to a gene are merged and the total number of non-redundant unique reads in the resulting merged exonic region is counted. The resulting number of reads is normalized with respect to the total size of merged exonic region of the gene and the total number of genomic unique and non-redundant reads in the particular sample, and the RPKM value is computed. The use of merged regions to compute read counts avoids the problem of double counting in regions where exons overlap with each other.
Using the RPKM values, the correlation coefficient (r) among the technical replicates for each sample was calculated, and this coefficient showed very little variation (R2 > 0.966) in all samples, consistent with the idea that the RNA-seq method is highly reproducible. Thus, data from all technical replicates for each sample were combined for further analysis.
ChIP-seq procedure
We dissected 200 pairs of bam testes in cold PBS and grouped in 200 μl PBS that contained protease inhibitor (Roche complete mini, cat. no. 11836153001) and 0.5 mM PMSF (MP Biomedicals, cat. no. 195381). We then added 5.5 μl of 37% fresh formaldehyde (Supelco, cat. no. 47083-U) and incubated at 37 °C for 15 min. The testes were spin at 2 000 r.p.m. for 2 min and washed two times with 450 μl cold 1× PBS (with inhibitors and PMSF). Then 200 μl lysis buffer (50 mM Tris-HCl (pH 7.6), 1 mM CaCl2, 0.2% Triton X-100, 5 mM butyrate, 1× proteinase inhibitor cocktail, and 0.5 mM fresh PMSF) was added and the tissues were homogenized thoroughly followed by incubation at room temperature (RT) for 10 min. We then carried out sonication with Microtip (Misonix Inc, Microson XL-2000) under the following procedure: 5′′ at power 20, rest for 50′′, 3-4 times, followed by spinning at 14 000 r.p.m. for 10′ at 4 °C. The chromatin was diluted 10× with RIPA buffer (10 mM Tris (pH 7.6), 1 mM EDTA, 0.1% SDS, 0.1% Na-Deoxycholate, and 1% Triton X-100, with protease inhibitors and PMSF) and 50 μl of this dilution was taken out as input.
We washed 40 μl of Dynabeads Protein A (Dynal Biotech ASA, Oslo, Norway) once with 1× PBS. We then added 4 μg of anti-RNA Pol II antibody (Abcam, ab5408) to the Dynabeads and incubated at RT for 40 min, followed by washing with 1× PBS. Next, 1 ml of the chromatin extract was added to the beads and the mixture was rotated at 4 °C overnight. Subsequent washing was performed as per the manufacturer's instructions. The beads were suspended in 100 μl of 1× TE containing 3 μl of 10% SDS and 5 μl of 20 mg/ml proteinase K. After overnight incubation at 65 °C, the supernatant was transferred to a new tube using a magnet (Dynal MPC-S) to precipitate the Dynabeads. Samples were treated by phenol/chloroform extraction, salt/EtOH precipitation, and resuspended in 50 μl 1× TE. The products were processed for Solexa sequencing according to the established protocol 95.
Comparison of RNA-seq with ChIP-seq
All Drosophila Ensembl genes were classified into five groups according to their RPKM value: 8 943 genes that have at least one RPKM were classified into high (2 981 genes, 41.07 < RPKM < 5 307), moderate (2 981 genes, 15.36 < RPKM < 41.06), or low (2 981 genes, 1.00 < RPKM < 15.36). 2 294 genes whose RPKM values were between 0.01 and 1.00 were considered as uncertain group and the rest 1 902 genes were classified as a silent group (RPKM = 0). The genes in each group were aligned to their transcription start site (TSS), using UCSC annotation (ftp://hgdownload.cse.ucsc.edu/goldenPath/dm3/database/). The read density was calculated in 5 bp windows.
Comparison of RNA-seq data with published microarray data
We first downloaded the CEL files generated by Parisi et al. 22 and Chintapalli et al. 96 from NCBI GEO database. We then extracted and normalized microarray signals with RMA function embedded in the limma package (downloaded from Bioconductor R packages (http://www.bioconductor.org)). Genes with multiple probes were filtered out if different probes give out inconsistent present (P) or absent (A) calls. Genes with at least three P calls or three A calls from four independent biological replicates for each sample were retained for further analysis. Differentially expressed genes were identified using the combination of P-value (P ≤ 0.05) and fold change (≥ 2) cutoffs, followed by comparison with their expression level using the RNA-seq data.
Heat map analyses
To visualize gene expression across different samples, heat map was generated using the TIGR software MeV v4.3.01 (http://www.tm4.org/mev.html). The hierarchical clustering was made using parameters of Euclidean distance and average linkage clustering. The sample columns were fixed and the genes were clustered with the optimized gene leaf order.
In situ hybridization
PCR primers were appended with a T7 RNA polymerase-binding site at their 5′ ends (5′-AAG GAT CCTAATACGACTCAC TATAGG GAG A-3′). For each of the following genes, we design two sets of primers to synthesize both sense (S) and antisense (AS) riboprobes. The primers' sequences are as follows:
T7_AS_trx, 5′-AAG GAT CCT AAT ACG ACT CAC TAT AGG GAG AAG GTC TCC TTG CCA AGC TTC AGA T-3′; S_trx, 5′-TAG AAA CGT GCT GGA GAC AAG CGA-3′; T7_S_trx, 5′-AAG GAT CCT AAT ACG ACT CAC TAT AGG GAG ATA GAA ACG TGC TGG AGA CAA GCG A-3′; AS_trx, 5′-AGG TCT CCT TGC CAA GCT TCA GAT-3′; T7_AS_lid, 5′-AAG GAT CCT AAT ACG ACT CAC TAT AGG GAG ACG CCA CTA TTG CTG TTG CTA TTG G-3′; S_lid, 5′-TCA AGA AGC GAT TAT GGC GCA GCA-3′; T7_S_lid, 5′-AAG GAT CCT AAT ACG ACT CAC TAT AGG GAG ATC AAG AAG CGA TTA TGG CGC AGC A-3′; AS_lid, 5′-CGC CAC TAT TGC TGT TGC TAT TGG-3′; T7_AS_rpd3, 5′-AAG GAT CCT AAT ACG ACT CAC TAT AGG GAG ATG CGT TAT TCG CCA CAT TGG ATC G-3′; S_rpd3, 5′-ACA GCA ACA AGG CAT CCT CAG AGA-3′; T7_S_rpd3, 5′-AAG GAT CCT AAT ACG ACT CAC TAT AGG GAG AAC AGC AAC AAG GCA TCC TCA GAG A-3′; AS_rpd3, 5′-TGC GTT ATT CGC CAC ATT GGA TCG-3′; T7_AS_Pcaf, 5′-AAG GAT CCT AAT ACG ACT CAC TAT AGG GAG AAC GTT CTC ATC CCG CGA CAC ATT A-3′; S_Pcaf, 5′-AAG GAT GAT TCG CCC ATC TGG GAT-3′; T7_S_Pcaf, 5′-AAG GAT CCT AAT ACG ACT CAC TAT AGG GAG AAA GGA TGA TTC GCC CAT CTG GGA T-3′; AS_Pcaf, 5′-ACG TTC TCA TCC CGC GAC ACA TTA-3′.
After the PCR amplification (Fermentas PCR purification Kit, cat no. K0702), we synthesize the RNA probes using DIG-labeled NTPs (Roche, cat no. 11277073910) and the T7 RNA polymerase (Roche, cat no. 10881767001) at 37 °C for 2 h, followed by hydrolysis with carbonate buffer (120 mM Na2CO3, 80 mM NaHCO3 (pH 10.2)) at 65 °C for 10 min. Whole mount testes in situ hybridization was carried out as previously described 97 with the following modifications: for each reaction, ∼15 pairs of testes were dissected from the y, w males that were less than 1 day post-eclosion. RNA hybridization to DIG-labeled probes was carried out in RNase-free tubes at 65 °C for 16-18 h. And the testes were subsequently washed six times (15 min each time) with 500 μl Hybe B (10.75 ml DEPC H2O, 25 ml Formamide (Fermentas, cat no. BP227500), 12.5 ml 20× SSC, 1 ml sheared herring sperm DNA (5 mg/ml, Sigma, cat no. D3159-10G), 250 μl heparin (10 mg/ml, Sigma, cat no. H3393-100KU), and 500 μl Tween 20 (Promega, cat no. H5151)) in a 65 °C water bath, followed by 15 min with each of the following solutions at RT: Hybe B:PBST (4:1); Hybe B:PBST (3:2); Hybe B:PBST (2:3); Hybe B:PBST (1:4); and 2× 15 min with 500 μl PBST (1× PBS plus 0.1% Tween-20).
The results were next developed using anti-digoxygenin antibody (Roche, cat no. 11093274910, at a 1:2 000 dilution) in RNase-free tubes at 4 °C overnight. The testes were then washed 4× 20 min with 500 μl PBST at RT, followed by 3× 5 min washes in freshly prepared NMTT (for 10 ml NMTT: 8.3 ml DEPC H2O, 1 ml Tri-HCl (pH 9.0), 200 μl of 5M NaCl, 500 μl of 1M MgCl2, and 10 μl Tween 20) at RT. The testes were then transferred from the tubes into the wells of dissecting dish. The NMTT buffer in each well was replaced by 300 μl NBT (4-nitro blue tetrazolium chloride) staining solution (298 μl NMTT, 1.35 μl NBT (Roche, cat. no. 11383213001) and 1.05 μl X-phosphate (Roche, cat. no. 11383221001)). The color reaction was allowed until differential staining signal could be detected in testes stained with antisense probe vs sense probe. The reaction was then stopped by 3× quick washes using PBST, 1× quick wash using 100% EtOH:PBST (1:1), 2× quick washes using 100% EtOH, and 2× quick washes using 100% EtOH:methyl salicylate (1:1). Approximately 300 μl GMM (3 ml methyl salicylate (Fermentas, cat. no. 03695-500) and 12 ml Canada balsam (Fermentas, cat. no. B10-100)) was added to the testes followed by overnight shaking at RT. Finally, the testes were mounted on slides and visualized using the CCD camera. The images were processed by Adobe Photoshop.
Transcript isoform expression from RNA-seq data
As many transcripts overlap with each other, not all isoforms are unambiguously detectable using RNA-seq short reads. To facilitate transcript isoform detection, we introduce the following natural ID system for transcripts. For each transcript T, let RT be the union of its exonic region that does not overlap with the rest of Ensembl transcripts. RT uniquely identifies transcript T. We then called a transcript T with a non-empty RT a uniquely detectable isoform. However, not all transcripts possess a non-empty R, because some are fully overlapped with other transcripts. We estimate the expression level of each uniquely detectable transcript isoform T by counting the number of unique and non-redundant reads in the RT region, followed by using the total size of RT as the normalization factor to compute the RPKM value. We used RPKM ≥ 1 as the cutoff for positive transcription.
We did not use exon-exon junction reads data to estimate the expression levels of alternatively spliced isoforms. The estimates of expression level using exon-exon junction read counts are not reliable due to a high level of sampling noise present at the small junctions. The relative sampling noise level is of the order n−1/2. The n is the expected number of reads at an exon-exon junction, which is dependent on the expression level of the corresponding transcript.
Entropy as a measure of alternatively spliced gene complexity
We set T1,T2,...,Tn to be the set of uniquely detectable isoforms of a gene; and x1,x2,...,xn to be the corresponding RPKM values estimated as described above. If only a few isoforms of a multi-isoform gene are expressed, the gene has low AS complexity. If majority of isoforms are expressed, the gene has high AS complexity. As a measure of AS complexity, we used gene entropy (or “S” in abbreviation) 65. The probability pk of isoform Tk is estimated as the frequency pk = xk/N, where N is the sum x1+x2+...+xn of isoforms' RPKM values of the corresponding gene. We added a pseudo-count ɛ = 0.001 to each xk to avoid zeroes. Using the pseudo-count-adjusted probability estimates, we computed the entropy of the gene as S = −∑ pk log2 pk.
By definition, the entropy is non-negative (S ≥ 0). It is zero if and only if all pk, except one, are zero. This is a desired property because if only one isoform of a multi-isoform gene is expressed, then the AS complexity of the gene should be zero. Another property of entropy is that it only depends on the probabilities (estimated as frequencies), but not on absolute RPKM values, as long as these values are large enough for a reliable estimate of frequencies. Thus, for a sufficiently expressed gene, entropy does not depend on the overall expression level of the gene. Therefore, it is meaningful to compare gene entropies across different samples. Entropy estimates for genes expressed at very low levels are less reliable. However, since we are mainly interested in the comparison of the overall AS complexity of a large number of genes across different samples, the effect of unreliable entropy estimates for a small subset of genes is insignificant.
The largest possible entropy log2 k for a gene with k uniquely detectable isoforms will be achieved when the frequencies of all isoforms are equal to 1/k. We normalized gene entropy by dividing it with log2 k. Therefore, the normalized entropy Snorm is always within the range from zero to one (0 ≤ Snorm ≤ 1).
Cumulative distribution function (CDF)
For each normalized entropy value x, the function CDF(x) gave out the fraction of genes whose normalized entropies are smaller than x. E.g., CDF (0.1) = 0.9 means that 90% of genes have normalized entropy less than 0.1. To statistically test if normalized entropies of AS loci in one sample is significantly larger than those in another sample, we used one-sided Kolmogorov-Smirnov test, which gave out P values shown in Figure 3B.
Differential splicing analysis
In Ensembl 50 database, 3 463 Drosophila genes (∼23%) have multiple isoforms. Among them, 2 094 were identified in our dataset that have more than one uniquely detectable isoform.
To visually display differential splicing of genes in two samples, we used the following method. For each gene, we asked the following questions: (1) Does it have transcript isoforms expressed both in samples 1 and 2? (2) Does it have transcript isoforms expressed only in sample 1? (3) Does it have transcript isoforms expressed only in sample 2?
The answer to each of these questions is yes (Y) or no (N). Thus, for each gene, there can be eight combinations of answers: YYY, YNY, NYY, YYN, NNY, YNN, NYN, NNN. In Figure 4A and 4D, Y is denoted by red color and N is denoted by blue color (The NNN category for each comparisons were not shown.).
We generated the list of candidate genes that undergo alternative splicing in a sex-specific or sex-biased manner in Table 3 as follows. The number of observed RNA-seq reads from a small exonic region is subjected to large sampling noise. The relative sampling noise level is of the order n−1/2. The n is the expected number of reads at the exonic region, which is dependent on the expression level of the corresponding transcript. To avoid the noise and thus false positive differentially spliced genes, we removed all uniquely detectable transcript isoforms with the ID region R of a total size smaller than 100 bp. Only genes with two or more uniquely detectable long R isoforms (UDLRI) were retained for further analysis. A differentially spliced gene must: (1) be sufficiently expressed in both samples (i.e., most expressed UDLRI isoforms have RPKM ≥ 10), (2) contain at least one ULDRI isoform that is not expressed (RPKM < 2) in one sample, but highly expressed (≥ 10 RPKM) in the other sample. The differentially spliced genes identified using the above procedure were then manually inspected on the UCSC genome browser to remove false positives and listed in Table 3 and Supplementary information, Table S5.
Chromosomal mapping assay
Chromosomal mapping was carried out with Pearson's chi-square test in R programming environment (R version 2.5.0, download from http://www.r-project.org). For bam testis vs WT testis comparison in Figure 6, we calculated the P-value in 2 × 2 tables with the gene numbers of the following categories: differentially expressed genes (≥ 2 fold change between bam testis and WT testis) on a certain chromosome arm (e.g., chrX), non-differentially expressed genes on a certain chromosome arm (e.g., chrX), differentially expressed genes on the other four chromosome arms (e.g., chr2L, chr2R, chr3L, and chr3R in together), and non-differentially expressed genes on the other four chromosome arms. The P < 0.01 was used to evaluate the significance of differential chromosomal distribution.
Identification of complementary-to-flybase transcribed regions (CFTRs)
The identification of CFTRs was modified from Barski et al. 95. Reads that can be mapped to known exons were all removed. Known exons were obtained from Ensembl database (http://www.ensembl.org/index.html) and the newest Flybase version (r5.19) (http://flybase.org/). The remaining reads were analyzed to discover CFTRs. For this purpose, CFTRs were defined for each sample by first calculating the number of reads aligning to each 40-bp window across the genome, and windows passing a P-value threshold (Poisson) of 0.05 were retained. Consecutive windows were grouped to form a large region, allowing a gap of two windows that did not satisfy the P-value cutoff. Finally, all CFTRs smaller than 100 bp (to reduce false positive caused by non-specific PCR amplification in some small regions that have high density of sequencing reads) or those contain fewer than 10 reads were eliminated. The RPKM for a CFTR was calculated according to their reads, sizes, and the total unique sequencing reads in each sample.
CFTRs were mapped to intergenic or intronic regions according to their mapped genomic loci (Ensembl). To eliminate possible contamination by pre-mRNA, intronic CFTRs were removed if their RPKM value was less than 10% of the RPKM of neighboring gene(s).
To validate the CFTRs, we compared them with the EST and mRNA sequences retrieved from UCSC or NCBI database, and genes predicted by N-SCAN (http://mblab.wustl.edu/predictions/Drosophila/dm3/) or CONTRAST (http://contra.stanford.edu/contrast/dm3.html). We found ∼14-23% CFTRs in each sample overlapped with these predicted genes (≥ 20 bp) at their corresponding genomic loci.
BLAST search
To determine potential products encoded by CFTR transcripts (e.g., protein coding sequences or non-coding RNA sequences), all CFTR sequences were extracted with a Perl script based on BDGP v5.0 and analyzed by BLASTX search against all non-redundant protein sequence with entries from GenPept, Swissprot, PIR, PDF, PDB, and NCBI RefSeq (4 October 2007) downloaded from ftp://ftp.ncbi.nih.gov/blast/db (24 December 2008). BLASTX were performed with local Linux system and the cutoffs were as follows: minimum score = 50, e-value ≤ 10−6, percent identity ≥ 50%, and have a minimum match of 30 amino acids.
Box plot analysis
The distribution of gene expression level was analyzed using boxplot in the R programming environment (R version 2.5.0, downloaded from http://www.r-project.org). The box represents the 25th and 75th percentiles, with the 50th percentile as a black bar. The whiskers refer to the outliers that are at least 1.5× IQR (interquartile range) from the box. The y axis represents the log2RPKM value.
Real-time RT-PCR validation
Real-time RT-PCR was used to validate sample-specific isoforms in all four pair-wise comparisons. In each experiment, two biological samples were prepared independently to carry out the real-time PCR experiments. For each PCR reaction, we run duplicates using SYBR Green PCR Master Mix (Fermentas, cat. no. K0221) in an ABI 7300 system. The following primers were used to validate corresponding genes for their common regions (c) and isoform-specific regions: p53.cF, 5′-GGA GAA GCA AAG GAA CAC ACG CAA-3′; p53.cR, 5′-ACT CGA TTC CGC TGA ACA AGC TCT-3′; p53.RA-F, 5′-ATT CCG ATC CCG ATA CCT CCA CC-3′; p53.RA-R, 5′-CAG CCA ATG TCG TGG CAC AAA GAA-3′; p53.RB-F, 5′-CTC TGC AGA AAC TTC GTT GCC GAT-3′; p53.RB-R, 5′-GCG GAC ACA AAT CGC AAC TGC TAA-3′; rab14.cF, 5′-TCT GGA GAC CGC ACG CAA GAT TTA-3′; rab14.cR, 5′-TTT AGC ACG AGC ACT GAT CCT TGG-3′; rab14.RA-F, 5′-ATT GCA ATC GAA TTC CGC ACA GCC-3′; rab14.RA-R, 5′-TTT AGT CCA CCT TAG GGA GCG AAC-3′; rab14.RB-F, 5′-TAA GCA GCG ACT ACG GTT GGA CAT-3′; rab14.RB-R, 5′-TGC GCA CTT TGC TCA TCT TGA CAC-3′; exu.cF, 5′-ACT TGT CAC CTC CTG CTC CAA ACT-3′; exu.cR, 5′-TGC TCG AGC TTC TGG ACA GCT ATT-3′; exu.RA-F, 5′-ACG CCC ACC AGG ATA TAA TTA CCG-3′; exu.RA-R, 5′-AAA GCG AAA GAG CCC ATC GAA ACC-3′; exu.RB-F, 5′-ACG CCC ACC AGG ATA TAA TTA CCG-3′; exu.RB-R, 5′-ATC TAG TGA AAG CGG TTC GCG T-3′; imp.cF, 5′-CAT TTC GCT CTG CAC AAG AAT GCG-3′; imp.cR, 5′-TGT TGG TCT GAA CGG TGT CGA GTT-3′; imp.RA/RC/RB-F, 5′-CTG GCC GAC TGT TGA GTT TCT TTC-3′; imp.RA/RC/RB-R, 5′-GCA ATA ACT ACA ACA ACA CAC GGC T-3′; imp.RG/RH-F, 5′-AAC TTG GTT GTG CGT TGC GA-3′; imp.RG/RH-R, 5′-AAG GCC AAA GGA AAG GCG AAA GAC-3′.
Accession codes
References
Cinalli RM, Rangan P, Lehmann R . Germ cells are forever. Cell 2008; 132:559–562.
Fuller MT, Spradling AC . Male and female Drosophila germline stem cells: two versions of immortality. Science 2007; 316:402–404.
Kiger AA, Jones DL, Schulz C, Rogers MB, Fuller MT . Stem cell self-renewal specified by JAK-STAT activation in response to a support cell cue. Science 2001; 294:2542–2545.
Tulina N, Matunis E . Control of stem cell self-renewal in Drosophila spermatogenesis by JAK-STAT signaling. Science 2001; 294:2546–2549.
Yamashita YM, Jones DL, Fuller MT . Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science 2003; 301:1547–1550.
Yamashita YM, Mahowald AP, Perlin JR, Fuller MT . Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science 2007; 315:518–521.
Brawley C, Matunis E . Regeneration of male germline stem cells by spermatogonial dedifferentiation in vivo. Science 2004; 304:1331–1334.
Maines JZ, Park JK, Williams M, McKearin DM . Stonewalling Drosophila stem cell differentiation by epigenetic controls. Development 2007; 134:1471–1479.
Buszczak M, Paterno S, Spradling AC . Drosophila stem cells share a common requirement for the histone H2B ubiquitin protease scrawny. Science 2009; 323:248–251.
Kai T, Williams D, Spradling AC . The expression profile of purified Drosophila germline stem cells. Dev Biol 2005; 283:486–502.
Maniatis T, Tasic B . Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 2002; 418:236–243.
Kim E, Magen A, Ast G . Different levels of alternative splicing among eukaryotes. Nucleic Acids Res 2007; 35:125–131.
Zarkower D . Establishing sexual dimorphism: conservation amidst diversity? Nat Rev Genet 2001; 2:175–185.
McKeown M . Sex differentiation: the role of alternative splicing. Curr Opin Genet Dev 1992; 2:299–303.
Burtis KC, Baker BS . Drosophila doublesex gene controls somatic sexual differentiation by producing alternatively spliced mRNAs encoding related sex-specific polypeptides. Cell 1989; 56:997–1010.
Demir E, Dickson BJ . Fruitless splicing specifies male courtship behavior in Drosophila. Cell 2005; 121:785–794.
Nagoshi RN, McKeown M, Burtis KC, Belote JM, Baker BS . The control of alternative splicing at genes regulating sexual differentiation in D. melanogaster. Cell 1988; 53:229–236.
McIntyre LM, Bono LM, Genissel A, et al. Sex-specific expression of alternative transcripts in Drosophila. Genome Biol 2006; 7:R79.
Telonis-Scott M, Kopp A, Wayne ML, Nuzhdin SV, McIntyre LM . Sex-specific splicing in Drosophila: widespread occurrence, tissue specificity and evolutionary conservation. Genetics 2009; 181:421–434.
Gupta V, Parisi M, Sturgill D, et al. Global analysis of X-chromosome dosage compensation. J Biol 2006; 5:3.
Parisi M, Nuttall R, Naiman D, et al. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science 2003; 299:697–700.
Parisi M, Nuttall R, Edwards P, et al. A survey of ovary-, testis-, and soma-biased gene expression in Drosophila melanogaster adults. Genome Biol 2004; 5:R40.
Sturgill D, Zhang Y, Parisi M, Oliver B . Demasculinization of X chromosomes in the Drosophila genus. Nature 2007; 450:238–241.
Schena M, Shalon D, Davis RW, Brown PO . Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995; 270:467–470.
Velculescu VE, Zhang L, Vogelstein B, Kinzler KW . Serial analysis of gene expression. Science 1995; 270:484–487.
Wilhelm BT, Marguerat S, Watt S, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 2008; 453:1239–1243.
Nagalakshmi U, Wang Z, Waern K, et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008; 320:1344–1349.
Lister R, O'Malley RC, Tonti-Filippini J, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008; 133:523–536.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B . Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008; 5:621–628.
Sultan M, Schulz MH, Richard H, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008; 321:956–960.
Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y . RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 2008; 18:1509–1517.
Wang Z, Gerstein M, Snyder M . RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009; 10:57–63.
McKearin DM, Spradling AC . bag-of-marbles: a Drosophila gene required to initiate both male and female gametogenesis. Genes Dev 1990; 4:2242–2251.
Gonczy P, Matunis E, DiNardo S . bag-of-marbles and benign gonial cell neoplasm act in the germline to restrict proliferation during Drosophila spermatogenesis. Development 1997; 124:4361–4371.
Terry NA, Tulina N, Matunis E, DiNardo S . Novel regulators revealed by profiling Drosophila testis stem cells within their niche. Dev Biol 2006; 294:246–257.
Fuller MT . Spermatogenesis. In: Bate M, Martinez Arias A, eds. The Development of Drosophila melanogaster. Vol 1. Cold Spring Harbor: Cold Spring Harbor Press, 1993: 71–147.
Spradling AC . Developmental genetics of oogenesis. In: Bate M, Martinez Arias A, eds. The Development of Drosophila melanogaster. Vol. 1. Cold Spring Harbor: Cold Spring Harbor Press, 1993:1–70.
Arbeitman MN, Fleming AA, Siegal ML, Null BH, Baker BS . A genomic analysis of Drosophila somatic sexual differentiation and its regulation. Development 2004; 131:2007–2021.
Lebo MS, Sanders LE, Sun F, Arbeitman MN . Somatic, germline and sex hierarchy regulated gene expression during Drosophila metamorphosis. BMC Genomics 2009; 10:80.
Arbeitman MN, Furlong EE, Imam F, et al. Gene expression during the life cycle of Drosophila melanogaster. Science 2002; 297:2270–2275.
Rathke C, Baarends WM, Jayaramaiah-Raja S, et al. Transition from a nucleosome-based to a protamine-based chromatin configuration during spermiogenesis in Drosophila. J Cell Sci 2007; 120 (Pt 9):1689–1700.
Fuller MT . Genetic control of cell proliferation and differentiation in Drosophila spermatogenesis. Semin Cell Dev Biol 1998; 9:433–444.
Beall EL, Lewis PW, Bell M, et al. Discovery of tMAC: a Drosophila testis-specific meiotic arrest complex paralogous to Myb-Muv B. Genes Dev 2007; 21:904–919.
Chen X, Hiller M, Sancak Y, Fuller MT . Tissue-specific TAFs counteract Polycomb to turn on terminal differentiation. Science 2005; 310:869–872.
Hiller M, Chen X, Pringle MJ, et al. Testis-specific TAF homologs collaborate to control a tissue-specific transcription program. Development 2004; 131:5297–5308.
Xi R, Xie T . Stem cell self-renewal controlled by chromatin remodeling factors. Science 2005; 310:1487–1489.
Tamkun JW . The role of brahma and related proteins in transcription and development. Curr Opin Genet Dev 1995; 5:473–477.
Elfring LK, Deuring R, McCallum CM, Peterson CL, Tamkun JW . Identification and characterization of Drosophila relatives of the yeast transcriptional activator SNF2/SWI2. Mol Cell Biol 1994; 14:2225–2234.
Gdula DA, Sandaltzopoulos R, Tsukiyama T, Ossipow V, Wu C . Inorganic pyrophosphatase is a component of the Drosophila nucleosome remodeling factor complex. Genes Dev 1998; 12:3206–3216.
Wu C, Tsukiyama T, Gdula D, et al. ATP-dependent remodeling of chromatin. Cold Spring Harb Symp Quant Biol 1998; 63:525–534.
Orlando V . Polycomb, epigenomes, and control of cell identity. Cell 2003; 112:599–606.
Francis NJ, Kingston RE . Mechanisms of transcriptional memory. Nat Rev Mol Cell Biol 2001; 2:409–421.
Smith ST, Petruk S, Sedkov Y, et al. Modulation of heat shock gene expression by the TAC1 chromatin-modifying complex. Nat Cell Biol 2004; 6:162–167.
Lee N, Zhang J, Klose RJ, et al. The trithorax-group protein Lid is a histone H3 trimethyl-Lys4 demethylase. Nat Struct Mol Biol 2007; 14:341–343.
Secombe J, Li L, Carlos L, Eisenman RN . The trithorax group protein Lid is a trimethyl histone H3K4 demethylase required for dMyc-induced cell growth. Genes Dev 2007; 21:537–551.
Eissenberg JC, Shilatifard A . Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol 2010; 339:240–9.
Kuo MH, Allis CD . Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 1998; 20:615–626.
De Rubertis F, Kadosh D, Henchoz S, et al. The histone deacetylase RPD3 counteracts genomic silencing in Drosophila and yeast. Nature 1996; 384:589–591.
Gildea JJ, Lopez R, Shearn A . A screen for new trithorax group genes identified little imaginal discs, the Drosophila melanogaster homologue of human retinoblastoma binding protein 2. Genetics 2000; 156:645–663.
Wang Z, Zang C, Cui K, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009; 138:1019–1031.
Lee N, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y . The H3K4 demethylase lid associates with and inhibits histone deacetylase Rpd3. Mol Cell Biol 2009; 29:1401–1410.
Spradling AC, Nystul T, Lighthouse D, et al. Stem cells and their niches: integrated units that maintain Drosophila tissues. Cold Spring Harb Symp Quant Biol 2008; 73:49–57.
Cheng J, Turkel N, Hemati N, et al. Centrosome misorientation reduces stem cell division during ageing. Nature 2008; 456:599–604.
Sheng XR, Brawley CM, Matunis EL . Dedifferentiating spermatogonia outcompete somatic stem cells for niche occupancy in the Drosophila testis. Cell Stem Cell 2009; 5:191–203.
Ritchie W, Granjeaud S, Puthier D, Gautheret D . Entropy measures quantify global splicing disorders in cancer. PLoS Comput Biol 2008; 4:e1000011.
Yeo G, Holste D, Kreiman G, Burge CB . Variation in alternative splicing across human tissues. Genome Biol 2004; 5:R74.
Elliott DJ, Grellscheid SN . Alternative RNA splicing regulation in the testis. Reproduction 2006; 132:811–819.
Cline TW . The Drosophila sex determination signal: how do flies count to two? Trends Genet 1993; 9:385–390.
Casper A, Van Doren M . The control of sexual identity in the Drosophila germline. Development 2006; 133:2783–2791.
Wawersik M, Milutinovich A, Casper AL, et al. Somatic control of germline sexual development is mediated by the JAK/STAT pathway. Nature 2005; 436:563–567.
Ko LJ, Prives C . p53: puzzle and paradigm. Genes Dev 1996; 10:1054–1072.
Giaccia AJ, Kastan MB . The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev 1998; 12:2973–2983.
Yamada Y, Davis KD, Coffman CR . Programmed cell death of primordial germ cells in Drosophila is regulated by p53 and the Outsiders monocarboxylate transporter. Development 2008; 135:207–216.
Zhang J, Schulze KL, Hiesinger PR, et al. Thirty-one flavors of Drosophila rab proteins. Genetics 2007; 176:1307–1322.
Zhang J, Fonovic M, Suyama K, Bogyo M, Scott MP . Rab35 controls actin bundling by recruiting fascin as an effector protein. Science 2009; 325:1250–1254.
Lighthouse DV, Buszczak M, Spradling AC . New components of the Drosophila fusome suggest it plays novel roles in signaling and transport. Dev Biol 2008; 317:59–71.
Barberan-Soler S, Zahler AM . Alternative splicing regulation during C. elegans development: splicing factors as regulated targets. PLoS Genet 2008; 4:e1000001.
Johnstone O, Lasko P . Translational regulation and RNA localization in Drosophila oocytes and embryos. Annu Rev Genet 2001; 35:365–406.
Macdonald PM, Luk SK, Kilpatrick M . Protein encoded by the exuperantia gene is concentrated at sites of bicoid mRNA accumulation in Drosophila nurse cells but not in oocytes or embryos. Genes Dev 1991; 5:2455–2466.
Hazelrigg T, Watkins WS, Marcey D, et al. The exuperantia gene is required for Drosophila spermatogenesis as well as anteroposterior polarity of the developing oocyte, and encodes overlapping sex-specific transcripts. Genetics 1990; 126:607–617.
Hazelrigg T, Tu C . Sex-specific processing of the Drosophila exuperantia transcript is regulated in male germ cells by the tra-2 gene. Proc Natl Acad Sci USA 1994; 91:10752–10756.
Herold N, Will CL, Wolf E, et al. Conservation of the protein composition and electron microscopy structure of Drosophila melanogaster and human spliceosomal complexes. Mol Cell Biol 2009; 29:281–301.
Fabrizio JJ, Hickey CA, Stabrawa C, et al. Imp (IGF-II mRNA-binding protein) is expressed during spermatogenesis in Drosophila melanogaster. Fly (Austin) 2008; 2:47–52.
Khil PP, Smirnova NA, Romanienko PJ, Camerini-Otero RD . The mouse X chromosome is enriched for sex-biased genes not subject to selection by meiotic sex chromosome inactivation. Nat Genet 2004; 36:642–646.
Wang PJ, McCarrey JR, Yang F, Page DC . An abundance of X-linked genes expressed in spermatogonia. Nat Genet 2001; 27:422–426.
Wu CI, Xu EY . Sexual antagonism and X inactivation--the SAXI hypothesis. Trends Genet 2003; 19:243–247.
Hense W, Baines JF, Parsch J . X chromosome inactivation during Drosophila spermatogenesis. PLoS Biol 2007; 5:e273.
Brett D, Pospisil H, Valcarcel J, Reich J, Bork P . Alternative splicing and genome complexity. Nat Genet 2002; 30:29–30.
Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR . Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci USA 2004; 101:15974–15979.
Black DL . Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 2003; 72:291–336.
Boyer LA, Mathur D, Jaenisch R . Molecular control of pluripotency. Curr Opin Genet Dev 2006; 16:455–462.
Jaenisch R, Young R . Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 2008; 132:567–582.
Chen X . Stem cells: What can we learn from flies? Fly (Austin) 2008; 2:19–28.
Buszczak M, Spradling AC . Searching chromatin for stem cell identity. Cell 2006; 125:233–236.
Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823–837.
Chintapalli VR, Wang J, Dow JA . Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet 2007; 39:715–720.
White-Cooper H, Schafer MA, Alphey LS, Fuller MT . Transcriptional and post-transcriptional control mechanisms coordinate the onset of spermatid differentiation with meiosis I in Drosophila. Development 1998; 125:125–134.
Gan Q, Schones DE, Eun S, et al. Monovalent and unpoised status of most genes in undifferentiated cell-enriched Drosophila testis. Genome Biol 2010; 11:R42.
Klose RJ, Kallin EM, Zhang Y . JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet 2006; 7:715–727.
Allis CD, Berger SL, Cote J, et al. New nomenclature for chromatin-modifying enzymes. Cell 2007; 131:633–636.
Wang L, Charroux B, Kerridge S, Tsai CC . Atrophin recruits HDAC1/2 and G9a to modify histone H3K9 and to determine cell fates. EMBO Rep 2008; 9:555–562.
Martin C, Zhang Y . The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 2005; 6:838–849.
Foglietti C, Filocamo G, Cundari E, et al. Dissecting the biological functions of Drosophila histone deacetylases by RNA interference and transcriptional profiling. J Biol Chem 2006; 281:17968–17976.
Zhu X, Singh N, Donnelly C, Boimel P, Elefant F . The cloning and characterization of the histone acetyltransferase human homolog Dmel\\TIP60 in Drosophila melanogaster: Dmel\\TIP60 is essential for multicellular development. Genetics 2007; 175:1229–1240.
Ciurciu A, Komonyi O, Pankotai T, Boros IM . The Drosophila histone acetyltransferase Gcn5 and transcriptional adaptor Ada2a are involved in nucleosomal histone H4 acetylation. Mol Cell Biol 2006; 26:9413–9423.
Zeremski M, Stricker JR, Fischer D, Zusman SB, Cohen D . Histone deacetylase dHDAC4 is involved in segmentation of the Drosophila embryo and is regulated by gap and pair-rule genes. Genesis 2003; 35:31–38.
Rudolph T, Yonezawa M, Lein S, et al. Heterochromatin formation in Drosophila is initiated through active removal of H3K4 methylation by the LSD1 homolog SU(VAR)3–3. Mol Cell 2007; 26:103–115.
Acknowledgements
We would like to thank Drs Allan Spradling (Carnegie Institution, USA), Karen Beemon and Mark Van Doren (The Johns Hopkins University, USA), and Chen lab members for critical readings and suggestions on the manuscript. We thank Dr Dustin E Schones (National Institutes of Health, USA) for help to set up the initial data analysis pipeline, and Caitlin Choi and Ankit Vartak for technical assistance with the PCR experiments. This work is supported in part by Research Grant No. 05-FY09-88 from the March of Dimes Foundation, the R00HD055052 NIH Pathway to Independence Award from NICHD, the 49th Mallinckrodt Scholar Award from the Edward Mallinckrodt, Jr. Foundation, support from the Johns Hopkins University (XC) and the Division of Intramural Research, the National Heart, Lung and Blood Institute, NIH (KZ).
Author information
Authors and Affiliations
Corresponding author
Additional information
(Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Figure S1
Methods of RNA-seq. (PDF 69 kb)
Figure S2
Comparison of RNA-seq data with microarray data. (PDF 83 kb)
Figure S3
More examples of genes that have sex-specific isoforms. (PDF 107 kb)
Figure S4
Venn diagrams of pair-wise comparisons of CFTRs. (PDF 96 kb)
Table S1
The RPKM values for expressed genes in at least one of the four samples. (XLS 2071 kb)
Table S2
Expression of genes involved in splicing in bam vs. wt testis. (XLS 89 kb)
Table S3
Meta table of multi-isoform genes in all four samples. (XLS 511 kb)
Table S4-1
Genes with different isoforms in bam testis vs. bam ovary samples. (XLS 260 kb)
Table S4-2
Genes with different isoforms in wt testis vs. wt ovary samples (XLS 260 kb)
Table S4-3
Genes with different isoforms in wt testis vs. bam testis samples (XLS 260 kb)
Table S4-4
Genes with different isoforms in wt ovary vs. bam ovary samples (XLS 260 kb)
Table S5
Genes that exhibit stage-specific or stage-biased isoforms in bam vs. wt gonads. (XLS 34 kb)
Table S6
Detection of CFTRs in all four samples. (XLS 1564 kb)
Rights and permissions
About this article
Cite this article
Gan, Q., Chepelev, I., Wei, G. et al. Dynamic regulation of alternative splicing and chromatin structure in Drosophila gonads revealed by RNA-seq. Cell Res 20, 763–783 (2010). https://doi.org/10.1038/cr.2010.64
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2010.64
Keywords
This article is cited by
-
The roles of sexual selection and sexual conflict in shaping patterns of genome and transcriptome variation
Nature Ecology & Evolution (2023)
-
Male-biased protein expression in primordial germ cells, identified through a comparative study of UAS vectors in Drosophila
Scientific Reports (2021)
-
Analysis of Drosophila melanogaster testis transcriptome
BMC Genomics (2018)
-
Enhancer of polycomb maintains germline activity and genome integrity in Drosophila testis
Cell Death & Differentiation (2018)
-
Sialotranscriptomics of Rhipicephalus zambeziensis reveals intricate expression profiles of secretory proteins and suggests tight temporal transcriptional regulation during blood-feeding
Parasites & Vectors (2017)
