
Abstract
The completion of genome sequences and subsequent high-throughput mapping of molecular networks have allowed us to study biology from the network perspective. Experimental, statistical and mathematical modeling approaches have been employed to study the structure, function and dynamics of molecular networks, and begin to reveal important links of various network properties to the functions of the biological systems. In agreement with these functional links, evolutionary selection of a network is apparently based on the function, rather than directly on the structure of the network. Dynamic modularity is one of the prominent features of molecular networks. Taking advantage of such a feature may simplify network-based biological studies through construction of process-specific modular networks and provide functional and mechanistic insights linking genotypic variations to complex traits or diseases, which is likely to be a key approach in the next wave of understanding complex human diseases. With the development of ready-to-use network analysis and modeling tools the networks approaches will be infused into everyday biological research in the near future.
Similar content being viewed by others

Introduction
To understand biological processes beyond single gene analyses, molecular biologists and geneticists often examine biomolecules in the framework of pathways and networks. This way the relationships of the molecules and the logic of operations can be readily captured and visualized. Cell cycle pathways, for example, have been beautifully delineated by yeast genetic analyses. However, no pathway is isolated. Even the fairly complete cell cycle pathway has by no means been comprehensively captured. Instead, cell cycle pathways are interconnected to many other cell processes through yet unknown mechanisms, for instance, their coupling to metabolism 1 and cellular growth and differentiation 2.
In the cell biomolecules are all interconnected with each other in a network, whose nodes are bio-molecules (proteins, genes, metabolites, and soon miRNAs) and whose edges are functional relationships among the nodes. Functional relationships include transcriptional and translational regulation, protein interactions, gene modifications, protein modifications, metabolic reactions, and indirect interactions like genetic interactions (e.g. synthetic lethality). The structure of these networks can vary over time and space constituting 'network dynamics' 3, 4. Although 'molecular networks', 'genetic networks' or 'gene networks' are sometimes used interchangeably, genetic or gene networks are actually the genetic templates for molecular networks, since interactions among gene products or between gene products and regulatory sequences are encoded by the genomic sequences (Figure 1).

Genetic network encodes molecular network, which in turn determines phenotypes. Although genes (except the regulatory sequences of the genes) do not directly participate in the molecular interactions, the potential interaction patterns (or what the molecular network is capable of doing) are encoded in genomic DNAs, and inherited from generation to generation. This concept is illustrated by the projections from genetic interactions to various types of molecular interactions between gene products, or between gene products and gene regulatory elements. However, both the genetic template and the actual steady states of the molecular interaction network are subject to change by environmental factors and developmental histories. Inside a system, the steady states of the molecular network directly correspond to the functional phenotype of the system, be it an organism, a tissue in an organisms, or other functional unit. When the changes affect the germlines of an organism, the changes become inheritable and play a role in evolution.
Why network biology?
Moving away from the popular Mendelian 'one gene - one phenotype' relationship, C H Waddington in 1957 came up with 'epigenetic landscape' to visually illustrate the multi-gene or network effects of genes on shaping the landscapes (various states) of cellular metabolism 5. Within these states canalization occurs through transition between the transiently stable states to final stable states. Waddington argued that it is these stable states and the transitions between them that determine developmental fates or phenotypes. Waddington did not know much about the material nature of genes; neither did he say anything about the molecular nature of cellular metabolism. Given current knowledge Waddington's landscapes model can be extended to incorporate molecular details. Translating 'cellular metabolism' as 'molecular networks' turns steady states into network representations or snapshots. Such steady states and the transition trajectories between them have been examined computationally through simulated genetic networks 6, 7, 8, and experimentally probed by gene expression profiling, such as profiles examining proliferation to differentiation transitions, gene mutation perturbations, or environmental or physiological challenges 9, 10, 11. The transition from one stable state to another is usually physiological but can be pathological, such as normal to diabetes mellitus, or normal to cancerous proliferation.
More explicitly, genetic networks are inheritable and thus subject to evolutionary selections and encode molecular networks (Figure 1). The landscape or dynamics of molecular networks can be revealed through their molecular signatures, such as conditional connectivity and gene expression profiles, which can be viewed as the molecular phenotypes of the networks 6, 12 (Figure 1). The functional phenotypes, such as growth rate of an organism, is the result of the molecular phenotypes, and often correspond to the steady states of the molecular networks 13 (Figure 1). Environmental factors and developmental history of an organism can alter both the genetic and molecular networks and the information transfer between them (Figure 1).
Not only the function of the gene products, but also their communication with each other, determines the function of the network and the selective advantage of the genes (Figure 1). Signal transduction research has been concerned with both function and communication. In this sense, network biology has been practiced for the last two decades, but the networks studied were often limited. Now with full gene lists available for many genomes, researchers can look beyond the few well-known genes to probe a larger unbiased network and address research questions in a larger context.
Along with the change in context, a change in research methodology is needed. Because mapping gene relationships entails a myriad of repetitive, labor-intensive experiments, improved high-throughput and automated technologies are needed; the fact that high-throughput experiments generate a sea of data that can no longer be catalogued or processed by hand means greater reliance on computer technologies; because high-throughput datasets can be filled with technical and biological noise and have different technical biases and coverage, improved statistics are needed to distinguish signal from noise, as well as better data integration to annotate the biologically relevant relationships; because the logical interpretation of the whole network is not easily comprehensible to the human brain, computational modeling is needed to predict the output from the signal input or system perturbation; because no analysis and modeling method is perfect, more detailed targeted biological experiments are needed to validate models before a hypothesis can be used to approach health and medical problems (Figure 2). Therefore network biology merits an independent research field because of different approaches, rather than different questions or conclusions. Although any approach has unique advantages and limitations, network biology as a new approach is bound to discover many properties that are not readily accessible by conventional molecular biology or genetics approaches.

Information flow in network biology research. Data or raw maps generated by large scale experiments first need to be annotated to extract biologically relevant information. Atop the annotated information, statistical analysis examines the design principles or dynamic parameters of the networks. Then, predictions can be made by applying the design principles and parameters in network models or simulations. Last but not least, the principles, parameters and predictions must be subjected to iterative experimental verification and refinement to approximate the true biological network as nearly as possible. With each step, data are churned or sublimed into information with a reduction in the amount of bits but an increase in accuracy, quality and usefulness.
Below I summarize the common methodologies, then review the information flow of network biology, with a focus on the dynamic modularity of the networks, and lastly take a look into the future (Figure 2).
Methods to acquire raw data for molecular networks
Small-scale gene-centric studies have delineated many valuable genetic and biochemical relationships between genes and pathways. This information forms the skeleton of the entire complete network and can serve as annotation standards. For now, such information covers only a tiny fraction of the full network and is biased towards certain biological functions. For example, protein interactions in the literature curated by HPRD (Human Protein Reference Database) 14 cover only 20 000 interactions out of a conservative estimate of 150 000 interactions and are strongly biased for cancer-related processes (Supplementary information, table S1). Large-scale experiments and data mining are obviously more effective for mapping genetic networks 15. Large-scale mapping approaches include: for transcription factor-target gene interactions, chromatin immunoprecipitation followed by microarray identification (ChIP-chip) or sequencing (ChIP-seq) and yeast one-hybrid screens; for protein-protein interactions (PPIs), co-affinity purification followed by mass spectrometry identification and yeast two-hybrid screens; and synthetic lethal screens for genetic interactions. Data mining efforts have been based on genomic sequences, such as gene fusion, gene neighborhood and phylogenic profiles 16, or on data generated by high-throughput experiments, such as correlated mRNA expression 17, 18, correlated phenotypic profiles 19, shared protein interaction partners 20, shared genetic interaction profiles 21, 22, shared subcellular localizations 23, or on a combination of genomic and experimental data, such as interolog and regulog predictions 24, 25.
Similar experimental and computational approaches have been used to generate small but more detailed maps for some specific biological processes, such as maps of the TGF-β pathway 26, 27, or C. elegans early embryogenesis 19. For further details on mapping molecular networks there are other more comprehensive reviews 15, 28, 29.
Inferring biological information by integration of raw data
How to use the sea of data to derive biological hypotheses is a most important and challenging task in the post-genomic era. Large-scale data are often noisy and cross-species annotation transfers are unreliable. Different interaction mapping techniques have dissimilar advantages and disadvantages, and often achieve limited coverage 30. Therefore, data integration is needed to obtain a more comprehensive, less technically biased and more accurate view of the true network. Integration approaches range from simple intersection analysis 18, 31, 32, to more sophisticated probability-based scoring systems where Bayesian probabilities are derived based on the number and strength of evidence an edge is associated with 33, 34, 35 (Figure 3).

An example of data integration by a probabilistic model. Heterogeneous dataset types can be evaluated by gold standard positive (GSP) and gold standard negative (GSN) functional relationships, for example PPIs. The potential of forming a true functional relationship can be scored as the likelihood ratio (LR) for protein/gene pairs to be true positive interactions versus true negative interactions, according to the GSP and GSN datasets. Taking each data type as independent, a Naïve Bayesian model can be used to integrate heterogeneous data. Each interaction is assigned a LR within a data type. When evidence arises from more than one dataset within a data type, the maximal LR among the datasets is used for a gene pair. Then the LRs given by different data types are multiplied to generate a final prediction score for a potential functional relationship. Based on an acceptable confidence level, a final integrated network can be obtained with each edge representing a likelihood of forming the functional relationship. PCC, GO, SSBP and DDI stand for Pearson Correlation Coefficient, Gene Ontology, Smallest Shared Biological Process and Domain-Domain Interaction, respectively. Adapted from 41.
Data integration can provide important guidance to downstream biological experimentation. Through integrating gene co-expression data, proteomics data and physical map of candidate disease loci, Mootha et al. narrowed a familial human cytochrome c oxidase deficiency down to a single gene and then validated the candidate gene by sequencing 36. By integrating transcriptome, interactome and phenome data, Gunsalus et al. identified molecular machines functioning in C. elegans early embryogenesis, and further inferred and experimentally verified the biological functions of ten previously uncharacterized proteins 19. By integrating anatomical expression, phenotype, functional annotation, microarray co-expression and PPI data from yeast, C. elegans and Drosophila using logistic regression analysis, Zhong and Sternberg predicted more than 18 000 genetic interactions, then experimentally validated the interactions for two human disease-related genes and identified 14 new modifier genes 37. By combining microarray analysis with interactome analysis, Gilchrist et al. found clues that ATF3 and Rel act downstream of a Toll-like receptor, then through further promoter analysis, experimental validation and kinetics modeling, they established a new regulatory circuit among the genes and downstream targets 38.
For small networks, where measurements based on system perturbations are plentiful, Bayesian networks 39 and correlation-based learning techniques 40 have been used to reverse engineer the active genetic networks. Based on thousands of single-cell data points measuring the level of 11 phosphoproteins and phospholipids upon activating or inhibiting each of them in human primary T cells, Bayesian network analysis accurately inferred the causal relationships among the 11 molecules 39. Based on the gene expression level changes upon overexpressing nine bacteria SOS pathway genes, multiple regression coefficients were successfully used to infer the key transcriptional regulations among the nine genes, which was further used to infer drug targets 40.
A recurring problem with data integration such as these is that once the integration is done, the results get out-of-date rapidly because of the continuous and ever-accelerating data generation from large-scale experiments. To solve this problem, a flexible integration method is needed to keep integrating new data as they are produced and updating the database. A Naïve Bayesian probability model is suitable for this purpose because it can easily integrate different types of biological data in different data formats. Using such a model, over 180 000 PPIs (protein-protein interactions) have been predicted among 10 000 human proteins based on 27 different 'omics' datasets, and deposited in the online database 'IntNetDB', which is intended to be updated whenever new datasets appear 41.
Uncovering network properties by statistical analyses
Networks have “emergent” properties that are distinct from those of their individual components. Emergent properties are non-linear, aggregated and combinatory effects generated by the interaction of the components of the networks. For example, properties such as topology, information flow and the stable states of a network can only be detected at the network level, not by examining the individual genes or proteins. The large number of nodes and edges in a network allow uncovering of network properties statistically in comparison to randomized or randomly generated networks. As the structural and dynamic features of genetic networks ultimately contribute to biological functions, robustness and evolvability 42 of these networks, a first step toward deciphering the design principles of genetic networks is to identify the physical and biological properties of the networks and, more importantly, the links between physical and biological properties.
Nearly all biological networks, including regulatory, interactome and metabolic networks, are scale-free networks. In a scale-free network most nodes have only one or two functional links, whereas a small number of nodes, the hubs, have many links. In scale-free networks the degree distribution (number of links per node) follows a power-law 43. Even though the scale-free topology of the marginally sampled networks may not represent the true topology of the complete networks 30, that hubs exist in these networks is unlikely to be derived randomly 30. “Degree centrality” has been linked to essentiality and conservation of genes 44, 45. Another network property of interest is node 'betweenness', the number of shortest paths between any pair of nodes passing through a node. “Betweenness centrality” better predicts the essentiality of a gene than degree centrality 46.
Networks of many different local topologies can all have the same degree distribution. Therefore the local topologies inform more about the details of the network structure. Most biological networks are small-world networks, that is, they tend to form local neighborhoods or communities as measured by the clustering coefficient 47, Jaccard index and cliquishness 20. The proteins in a local community tend to have similar functions and therefore likely correspond to a functional gene cluster or neighborhood or module 48. Various algorithms have been applied to find local communities based solely on the static structure of networks 49, 50, 51.
Certain local interaction patterns or “network motifs”, such as feedback and feed-forward loops, are enriched in molecular networks 52, 53. In regulatory networks, where the directions of the edges are identifiable, feedback and feed-forward loops are apparently over-represented compared to randomly generated networks of the same degree distributions 52. Similar networks can be identified based on their distinct signatures of enriched motifs. In other words, the appearance frequency of different motifs within each network is a characteristic shared by networks of the same type. For example, the regulatory networks of microorganisms and higher organisms can be clearly distinguished by the preference for feed-forward loops in the former and both feed-forward and feedback loops in the latter 54. The scale-free and small-world properties and network motifs have been reviewed extensively before 43.
Biological networks are not static; their functions are determined not only by their static structures but also by their dynamic structural rearrangements. Yeast gene regulatory networks have different properties under different conditions. Active networks under normal physiological conditions tend to have long average path length and are densely connected, whereas active networks under exogenous challenges tend to have short average path length and are less inter-connected 55. Analyses on signal propagation within a network composed of 23 ligand-induced signaling pathways in hippocampal CA1 neurons revealed that signals branch early in the network for ligands that induce rapid and transient changes, late for G protein-coupled receptors and little for those ligands that cause permanent changes. Negative and positive feedback loops are concentrated at early and late steps, respectively, suggesting a balancing barrier in the early stages and a persistent effect in the late stages of signaling. At 15 steps from the ligand-receptor interactions, nearly all the genes in the network are engaged, so any ligand ultimately affects the entire network 56.
Genetic networks differ from non-biological networks in that they are subject to evolutionary selection. First, genetic buffering may be important in accelerating evolution of the molecular networks. Simulated evolution of the gene network has shown that many single gene mutations or deletions can give rise to phenotype variations in the progeny. That is, many genes may have buffering effects towards genetic perturbations, while removing or mutating the genes can accelerate evolutionary adaptation 6. Second, environmental constraints are important determinants of the evolutionary direction of the molecular networks. For instance, given the nutrient environment of the symbiotic bacteria and the metabolic network of their ancestral free-living bacteria, the minimal metabolic network of these symbiotic bacteria can be predicted with fairly good accuracy 57. Thus, environmental constraints are pivotal to shape the steady states of the molecular networks (molecular phenotypes) and to select the corresponding genetic networks (genotypes) (Figure 1). Third, local functional units of the networks, such as protein complexes and signaling circuits, are likely evolutionarily selected and preserved. Indeed genes inside the same protein complexes and pathways are conserved across species 53, 58. However, within a single organism, the network motifs are mostly not conserved among different sets of paralogous genes. Network motifs in regulatory networks have been suggested to evolve by convergent evolution or divergence after gene duplication, because simple duplications of network motifs with paralogous genes in either E. coli or yeast gene regulatory networks are rare 59, 60. Instead, the whole genome duplication in yeast may have served to derive functional sub-networks under different regulatory controls 61, 62. Such results are naturally expected if the selections on network motifs within a species are driven by function rather than driven by structure.
Predicting network behavior by in silico modeling
A network can be represented in silico and its properties studied using computer-simulated perturbations. Several models have been successfully employed to predict the behavior of networks. The most successful is the flux balance model for metabolic networks. The model assumes that metabolic pathways are utilized to maximize the metabolites required under particular growth conditions. Flux balance optimizes metabolic flow for maximal biomass production of certain metabolites, for example ATP, under mass balance and physicochemical constraints of steady states to compute whole-cell metabolic fluxes and growth rates 63.
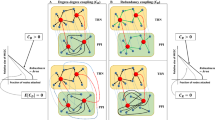
In principle, if a signaling network follows similar optimization rules and has known constraints, a constraints-based model can predict the output of the signaling network (Figure 4). However, signaling networks are more complex than metabolic networks, whose function is mainly energy generation and whose output is change in growth rate. Currently not enough is known about what principles guide the flow of information in signaling networks, or genetic networks in general, and what constraints limit the functions and outputs of such networks.

Constraint-based modeling. Constraint-based modeling has been successfully used in metabolic networks. In principle, if the constraints for signaling networks can be identified, such a model can be also applied to signaling networks. Stoichiometric, thermodynamic and capacity constraints are the major constraints for metabolic networks. Major constraints for signaling networks are probably temporal and spatial constraints, which have yet to be fully understood. (Modified based on 63 with permission from the authors and Proc Natl Acad Sci USA).
Network motifs can be viewed as building blocks of the networks, and are also small enough for accurate mathematic modeling and sometimes ex vivo reconstitution (a.k.a. 'synthetic biology') 64. When only a few network motifs are considered and kinetic parameters are well-measured, kinetic models have been successfully used to predict the signaling properties of small networks 4. For example, the function of the PKC (protein kinase C) mediated positive feedback loop and the function of MAPK phosphatase in tuning ligand sensitivity have been revealed through kinetic analyses of the MAP kinase signaling module 65. In similar scale networks, probability-based reverse-engineered network model can predict the qualitative instead of quantitative or dynamic outputs of networks 40. In small networks where the kinetic parameters are unknown, but all deterministic nodes and edges are known, Boolean network models have been used to predict outputs and steady states of the networks, such as the cell cycle network 8 and the segment polarity gene expression network in Drosophila embryo 66.
Today most kinetic parameters of the interactions are unknown and the eukaryotic cellular networks are far from being completely mapped, so these modeling approaches remain ambitious future projects for accurate systems modeling of multicellular organisms. The potential of such models to personalize medical treatment based on individual variations and to predict systems-level drug effects will drive biomedical and pharmaceutical research toward such systems models.
Building accurate and practically useful models needs to start with experimental observations. To test the validity of and refine the models or to get individualized parameters for the models needs iterative experimental probing of network features according to the predictions of the models 67, 68.
Dynamic modular networks for mathematical modeling
Mounting evidence points to a modular structure for the transcriptome and interactome. For example, transcriptionally co-regulated genes, or tightly interacting gene neighborhoods (cliquish clusters) in the interactome networks, often correspond to molecular machines and other functional modules 17, 69. Although some modules, such as stable protein complexes are constantly present in multiple cellular conditions, other functional modules are dynamically constructed and deconstructed. For instance, cell cycle dependent protein complexes are dynamically constructed and disassembled during different phases of the cell cycle 70. As another example, hubs in the yeast interactome network can be categorized as 'date' or 'party' hubs, where a date hub interacts with its partners at different times and a party hub interacts with its partners simultaneously. Date hubs tend to connect different functional modules, whereas party hubs tend to function within the modules, suggesting dynamically organized modularity in the yeast interactome network 31, 71. A network module has been defined as meeting three criteria: precisely defined inputs and outputs, comparable timescale, and spatial co-localization under certain condition 3. Such a definition covers both static and dynamic network modules with an emphasis on context-coherence.
As the genes or proteins inside a module are synchronously regulated and functionally insulated from other parts of the network, they can sometimes be regarded as a single functional unit, that is, one single node in the network 72. Such modules can be extracted from a static network through statistical analysis by examining the dynamic changes of the nodes and/or edges. For example, by examining a subnetwork of the PPIs between genes that have similar or opposite expression patterns across different human brain samples, or across different fruit fly populations, we could dissect PPI network modules where 1) the expression profiles of genes within a module are similar, 2) the PPIs between genes that have similar expression patterns are maximally enclosed within a module, and 3) the PPIs between genes that have opposite expression patterns are optimally distributed between modules. Two of the modules so found are transcriptionally anti-correlated modules at the tissue and organismal levels, and relate to cellular proliferation and differentiation states. The two modules reflect the temporal switch between two alternative states - proliferation and differentiation states 73. Computationally, the expression profiles of proliferation and differentiation modules found at tissue and organism levels can be explained by a simply toggle switch (Figure 5a). However, even with current limited network coverage, there are over 15 feedback control loops between the two modules. The PPIs connecting the two modules preferentially connect regulatory genes, especially oncogenes and tumor suppressor genes. It seems that to robustly coordinate and fine-tune the relationship between the two modules, nature has selected for greater redundancy and more complex regulatory circuitry than what is merely sufficient (Figure 5b) 73.

Regulatory circuitry between the proliferation and differentiation modules is more than merely sufficient. (A) Theoretically, the expression profiles of the differentiation (D) and proliferation (P) modules can be explained by a simple toggle switch, i.e., two mutually inhibitory interactions between the two modules. D and P modules are represented by two squares. Their mutually inhibitory relationships are represented by barbed lines. (B) In reality, the circuitry is more complex. Even at limited network coverage, many feedback loops can be seen to traverse between the two modules. Each node represents a gene. Solid edges represent directional protein interactions, and dashed edges represent transcriptional regulation. Red and green colors of edges represent transcriptional correlations and anti-correlations, respectively. Adapted from reference 73.
We have found that genes inside the aging-related subnetworks aggregate into several modules related to cellular temporal switches 74. Two modules of a switch not only correspond to two alternative cellular states, but also assume opposite expression changes during aging. Such modules are connected through concerted transcriptional regulation and extensive PPIs, with genes connecting these modules through PPIs more likely to affect network stability and organism lifespan 74.
Network complexity is incurred by robustness, fine tuning, spatial and temporal compartmentalization and dynamic reorganization between and within functional modules under different conditions 75. Such complexity makes it difficult to intuitively infer the output of a modular network. Therefore, computational modeling is necessary to simplify and dissect the circuitry required to achieve the observed functional states of the modules. With well-defined inputs and outputs of the modules, a module-level network will tremendously simplify the view of a cellular network and make it amenable to computational modeling. Modeling metabolic network at the modular level, epistatic relationships have been revealed for modules in these networks 76. When the complex regulatory network of pattern formation during Drosophila embryogenesis is reduced to two or three coarse grain modules, the functional output of the full regulatory network can still be robustly reproduced through computational simulation of the modular network. Thus modular network topology and kinetics are sufficient to capture the overall function of the network 77.
Synthetic biology can help reconstitute and verify modeling results. Many types of simple regulatory networks have been successfully constructed 64. The output of some of these networks has been demonstrated to be exactly as computationally predicted, whereas that of others is not yet fully predictable by computational modeling 78. Synthetic biology has unique advantages in formulating or predicting the modular interactions in a modular network. If a catalog of synthetic circuitry with accompanying dynamics is available, one can compare a dynamic profile at hand with such a catalog to find a possible minimal regulatory circuit generating the profile. Instead of going into the complex in vivo system to test various models and parameters for constructing a correct computational model, a synthetic 'model' biological network system provides a simpler middle ground.
In summary, modular network models may bridge the current gap between statistical analyses and computational modeling, overcome the computational hurdles imposed by the large sizes of systems-wide networks, and allow examination of dynamic outputs.
Dynamic network modularity links genotypes to complex phenotypes
A complex phenotype is usually determined by multiple genes, although the genes do not necessarily have the same influence over a phenotype. Traditional genetic linkage and genetic association tests are insufficient for complex diseases 79. Because of the weak effect of each individual gene, it is hard to distinguish a true genetic association from background noise. Even when an associative locus is found, it usually encloses a large genetic interval, often covering more than 100 genes 80, 81, making it hard to determine the molecular functions of the associated locus. Here enters network integration. The state of the molecular network that corresponds to the current overall phenotype of the system is determined by the gene network and its interactions with environmental and developmental factors (Figure 1). Genes contributing to a particular complex phenotype or a complex disease are usually connected to each other to form a subnetwork 19, 82. The genes within the subnetwork are more densely connected to each other than toward those outside of the subnetwork. A gene subnetwork usually contains a few functional modules, with genes inside the module having similar biological functions 19, 73.
Lage et al. have developed a Bayesian predictor using the phenotypes of the first degree interactors to narrow down the associative genes in large genomic intervals that are previously mapped as disease-associated loci. They obtained confident predictions for 91 of a total of 870 intervals 81. As this result is based on only the first degree interactors, including information from a disease-related subnetwork or modules may further increase the coverage. Using as query genes four genes known to be associated with breast cancer, Pujana et al. have identified a subnetwork of genes that are co-regulated with the query genes. After prioritizing the genes in the subnetwork according to the strength of the functional links to the query genes, a link between breast cancer susceptibility and centrosome dysfunction was found. A gene linking the two phenomena was found genetically associated with breast cancer occurrence in two independent patient populations 83. Ghazalpour et al. developed a new metric called modular QTL (mQTL) to evaluate if a quantitative trait locus (QTL) is enriched in a network module 84. Although their network included only co-expression relationships, the approach can be potentially adapted for networks composed of other types of 'omics' data.
A network approach can reveal not only which genes are associated with a complex disease, but also the genetic interactions, potential pathways and regulatory networks contributing to the disease. New integrative strategies based on network analysis are undoubtedly promising approaches to link genotypic variations to susceptibility for complex diseases.
An outlook
Network biology still faces many challenges. Datasets are both data-rich and data-poor, that is, false positives and limited coverage are still the rule. The transition from model organisms to human means magnitudes of increase in complexity of both experimentation and computation. Most edges in network maps are still missing the signs and directions. Post-transcriptional modifications cannot be easily monitored at large scale. Tissue and cell type specificities are not considered. Genome-wide dynamic measurements are costly. However, with development of novel high-throughput and single cell dynamic measurement techniques and with improvement of accuracy and coverage over existing technologies, high-throughput experiments will continue to accelerate data acquisition and raise further need for data processing, integration, analysis and modeling.
Physics and mathematics have provided important tools for network biology on data mining, network metrics and modeling techniques, and will be driven by new challenges and requirements of network biology to develop new network metrics and dynamic models. However, the change to network biology does not simply entail handing over biology to physicists and mathematicians. A good understanding of biology is needed to ask the right questions, to choose proper network analysis tools, and to confirm analysis results by solid experimentation. After all, network biology is biology. The fundamental goal of network biology is the same as molecular biology: to understand basic biological processes and the mechanisms of human diseases.
Since there is no need to understand how a computer works to use a computer, there is no need to be mathematicians or physicists to use network analysis and modeling approaches to study biology. In fact, many of the analysis and modeling approaches I've discussed have been implemented in ready-to-use and freely accessible databases and software. BIND, DIP, GRID, MINT, IntAct and KEGG are databases aiming to collect all the interactions and pathways for cellular networks. Cytoscape is a free and easy-to-use network visualization tool 85. TopNet is an online package that extracts standard network properties such as degree distribution, clustering coefficient and shortest distances 86. MCode is a program to extract highly clustered gene neighborhoods 69. MFinder and FANMOD are programs for finding over-represented network motifs 54, 87. Dizzy, BioNetS and RMBNToolbox are network kinetics simulators 88, 89, 90 (Table 1). The functions and performances of 12 different modeling tools have been compared in detail 91. Free, ready-to-use and user-friendly software will soon be available for other analysis and modeling approaches. Soon, I expect that biologists will be able to use these programs as they use molecular biology reagents and kits, or homology search tools like BLAST. Network-based interactive modeling and experimental approaches will then be the norm rather than a hope for modern biologists.
(Supplementary information is linked to the online version of the paper on the Cell Research website.)
References
Tu BP, Kudlicki A, Rowicka M, McKnight SL . Logic of the yeast metabolic cycle: temporal compartmentalization of cellular processes. Science 2005; 310:1152–1158.
Bjorklund M, Taipale M, Varjosalo M, et al. Identification of pathways regulating cell size and cell-cycle progression by RNAi. Nature 2006; 439:1009–1013.
Papin JA, Hunter T, Palsson BO, Subramaniam S . Reconstruction of cellular signalling networks and analysis of their properties. 2005; Nat Rev Mol Cell Biol 6:99–111.
Kholodenko BN . Cell-signalling dynamics in time and space. 2006; Nat Rev Mol Cell Biol 7:165–176.
Waddington CH . The Strategy of the Genes. London: Allen & Unwin. 1957.
Bergman A, Siegal ML . Evolutionary capacitance as a general feature of complex gene networks. Nature 2003; 424:549–552.
Kauffman SA . Metabolic stability and epigenesis in randomly constructed genetic nets. J Theor Biol 1969; 22:437–467.
Li F, Long T, Lu Y, Ouyang Q, Tang C . The yeast cell-cycle network is robustly designed. Proc Natl Acad Sci USA 2004; 101:4781–4786.
Chen JF, Mandel EM, Thomson JM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 2006; 38:228–233.
Huang S, Eichler G, Bar-Yam Y, Ingber DE . Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys Rev Lett 2005; 94:128701.
Jasper H, Benes V, Atzberger A, et al. A genomic switch at the transition from cell proliferation to terminal differentiation in the Drosophila eye. Dev Cell 2002; 3:511–521.
Hughes TR, Marton MJ, Jones AR, et al. Functional discovery via a compendium of expression profiles. Cell 2000; 102:109–126.
Papin JA, Reed JL, Palsson BO . Hierarchical thinking in network biology: the unbiased modularization of biochemical networks. Trends Biochem Sci 2004; 29:641–647.
Peri S, Navarro JD, Kristiansen TZ, et al. Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res 2004; 32:D497–D501.
Vidal MA . Biological atlas of functional maps. Cell 2001; 104:333–339.
Marcotte EM . Computational genetics: finding protein function by nonhomology methods. Curr Opin Struct Biol 2000; 10:359–365.
Eisen MB, Spellman PT, Brown PO, Botstein D . Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 1998; 95:14863–14868.
von Mering C, Krause R, Snel B, et al. Comparative assessment of large-scale data sets of protein-protein interactions. Nature 2002; 417:399–403.
Gunsalus KC, Ge H, Schetter AJ, et al. Predictive models of molecular machines involved in Caenorhabditis elegans early embryogenesis. Nature 2005; 436:861–865.
Goldberg DS, Roth FP . Assessing experimentally derived interactions in a small world. Proc Natl Acad Sci USA 2003; 100:4372–4376.
Tong AH, Lesage G, Bader GD, et al. Global mapping of the yeast genetic interaction network. Science 2004; 303:808–813.
Tong AH, Evangelista M, Parsons AB, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368.
Lin N, Wu B, Jansen R, Gerstein M, Zhao H . Information assessment on predicting protein-protein interactions. BMC Bioinformatics 2001; 5:154.
Matthews LR, Vaglio P, Reboul J, et al. Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or “interologs”. Genome Res 2001; 11:2120–2126.
Yu H, Luscombe NM, Lu HX, et al. Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res 2004; 14:1107–1118.
Barrios-Rodiles M, Brown KR, Ozdamar B, et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science 2005; 307:1621–1625.
Tewari M, Hu PJ, Ahn JS, et al. Systematic interactome mapping and genetic perturbation analysis of a C. elegans TGF-beta signaling network. Mol Cell 2004; 13:469–482.
Cusick ME, Klitgord N, Vidal M, Hill DE . Interactome: gateway into systems biology. Hum Mol Genet 2005; 14 Spec No. 2: R171–R181.
Ge H, Walhout AJ, Vidal M . Integrating 'omic' information: a bridge between genomics and systems biology. Trends Genet 2003 19:551–560.
Han JD, Dupuy D, Bertin N, Cusick ME, Vidal M . Effect of sampling on topology predictions of protein-protein interaction networks. Nat Biotechnol 2005; 23:839–844.
Han JD, Bertin N, Hao T, et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 2004; 430:88–93.
Said MR, Begley TJ, Oppenheim AV, Lauffenburger DA, Samson LD . Global network analysis of phenotypic effects: protein networks and toxicity modulation in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 2004; 101:18006–18011.
Jansen R, Yu H, Greenbaum D, et al. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science 2003; 302:449–453.
Lee I, Date SV, Adai AT, Marcotte EM . A probabilistic functional network of yeast genes. Science 2004; 306:1555–1558.
Rhodes DR, Tomlins SA, Varambally S, et al. Probabilistic model of the human protein-protein interaction network. Nat Biotechnol 2005; 23: 951–959.
Mootha VK, Lepage P, Miller K, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci USA 2003; 100:605–610.
Zhong W, Sternberg PW . Genome-wide prediction of C. elegans genetic interactions. Science 2006; 311:1481–1484.
Gilchrist M, Thorsson V, Li B, et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 2006; 441:173–178.
Sachs K, Perez O, Pe'er D, Lauffenburger DA, Nolan GP . Causal protein-signaling networks derived from multiparameter single-cell data. Science 2005; 308:523–529.
Gardner TS, di Bernardo D, Lorenz D, Collins JJ . Inferring genetic networks and identifying compound mode of action via expression profiling. Science 2003; 301:102–105.
Xia K, Dong D, Han JD . IntNetDB v1.0: An integrated protein-protein interaction network database generated by a probabilistic model. BMC Bioinformatics 2006; 7:508.
Kirschner M, Gerhart J . Evolvability. Proc Natl Acad Sci USA 1998; 95:8420–8427.
Barabasi AL, Oltvai ZN . Network biology: understanding the cell's functional organization. Nat Rev Genet 2004; 5:101–113.
Fraser HB, Hirsh AE, Steinmetz LM, Scharfe C, Feldman MW . Evolutionary rate in the protein interaction network. Science 2002; 296:750–752.
Jeong H, Mason SP, Barabasi AL, Oltvai ZN . Lethality and centrality in protein networks. Nature 2001; 411:41–42.
Yu H, Kim PM, Sprecher E, Trifonov V, Gerstein M . The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput Biol 2007; 3:e59.
Watts DJ, Strogatz SH . Collective dynamics of 'small-world' networks. Nature 1998; 393:440–442.
Hartwell LH, Hopfield JJ, Leibler S, Murray AW . From molecular to modular cell biology. Nature 1999; 402:C47–52.
Girvan M, Newman ME . Community structure in social and biological networks. Proc Natl Acad Sci USA 2002; 99:7821–7826.
Rives AW, Galitski T . Modular organization of cellular networks. Proc Natl Acad Sci USA 2003; 100:1128–1133.
Rung J, Schlitt T, Brazma A, Freivalds K, Vilo J . Building and analysing genome-wide gene disruption networks. Bioinformatics 2002; 18 Suppl 2:S202–S210.
Milo R, Shen-Orr S, Itzkovitz S, et al. Network motifs: simple building blocks of complex networks. Science 2002; 298:824–827.
Wuchty S, Oltvai ZN, Barabasi AL . Evolutionary conservation of motif constituents in the yeast protein interaction network. Nat Genet 2003; 35:176–179.
Milo R, Itzkovitz S, Kashtan N, et al. Superfamilies of evolved and designed networks. Science 2004; 303:1538–1542.
Luscombe NM, Babu MM, Yu H, et al. Genomic analysis of regulatory network dynamics reveals large topological changes. Nature 2004; 431:308–312.
Ma'ayan A, Jenkins SL, Neves S, et al. Formation of regulatory patterns during signal propagation in a Mammalian cellular network. Science 2005; 309:1078–1083.
Pal C, Papp B, Lercher MJ, et al. Chance and necessity in the evolution of minimal metabolic networks. Nature 2006 440:667–670.
Sharan R, Suthram S, Kelley RM, et al. Conserved patterns of protein interaction in multiple species. Proc Natl Acad Sci USA 2005; 102:1974–1979.
Conant GC, Wagner A . Convergent evolution of gene circuits. Nat Genet 2003; 34:264–266.
Teichmann SA, Babu MM . Gene regulatory network growth by duplication. Nat Genet 2004; 36:492–496.
Conant GC, Wolfe KH . Functional partitioning of yeast co-expression networks after genome duplication. PLoS Biol 2006; 4:e109.
Wapinski I, Pfeffer A, Friedman N, Regev A . Natural history and evolutionary principles of gene duplication in fungi. Nature 2007; 449:54–61.
Famili I, Forster J, Nielsen J, Palsson BO . Saccharomyces cerevisiae phenotypes can be predicted by using constraint-based analysis of a genome-scale reconstructed metabolic network. Proc Natl Acad Sci USA 2003; 100:13134–13139.
Hasty J, McMillen D, Collins JJ . Engineered gene circuits. Nature 2002; 420:224–230.
Bhalla US, Ram PT, Iyengar R . MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science 2002; 297:1018–1023.
Albert R, Othmer HG The topology of the regulatory interactions predicts the expression pattern of the segment polarity genes in Drosophila melanogaster. J Theor Biol 2003; 223:1–18.
Ideker T, Thorsson V, Ranish JA, et al. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 2001; 292:929–934.
King RD, Whelan KE, Jones FM, et al. Functional genomic hypothesis generation and experimentation by a robot scientist. Nature 2004; 427:247–252.
Bader GD, Hogue CW . An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003; 4:2.
de Lichtenberg U, Jensen LJ, Brunak S, Bork P Dynamic complex formation during the yeast cell cycle. Science 2005; 307:724–727.
Bertin N N.S, D. D, et al. Confirmation of organized modularity in the yeast interactome. PLoS Biology 2007; 5:e153.
de Bivort B, Huang S, Bar-Yam Y . Dynamics of cellular level function and regulation derived from murine expression array data. Proc Natl Acad Sci USA 2004; 101:17687–17692.
Xia K, Dong D, Xue H, et al. Identification of the Proliferation/Differentiation Switch in the Cellular Network of Multicellular Organisms. PLoS Computational Biology 2006; 2:e145.
Xue H, Xian B, Dong D, et al. A modular network model of aging. Mol Syst Biol 2007; 3:147.
Segal E, Friedman N, Kaminski N, Regev A, Koller D . From signatures to models: understanding cancer using microarrays. Nat Genet 2005; 37 Suppl:S38–S45.
Segre D, Deluna A, Church GM, Kishony R . Modular epistasis in yeast metabolism. Nat Genet 2005; 37:77–83.
Ma W, Lai L, Ouyang Q, Tang C . Robustness and modular design of the Drosophila segment polarity network. Mol Syst Biol 2006; 2:70.
Lahav G, Rosenfeld N, Sigal A, et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet 2004; 36:147–150.
Emahazion T, Feuk L, Jobs M, et al. SNP association studies in Alzheimer's disease highlight problems for complex disease analysis. Trends Genet 2001; 17:407–413.
Calvo S, Jain M, Xie X, et al. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat Genet 2006; 38:576–582.
Lage K, Karlberg EO, Storling ZM, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol 2007; 25:309–316.
Gandhi TK, Zhong J, Mathivanan S, et al. Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 2006; 38:285–293.
Pujana MA, Han JD, Starita LM, et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet 2007; 39:1338–1349.
Ghazalpour A, Doss S, Zhang B, et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet 2006; 2:e130.
Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13:2498–2504.
Yu H, Zhu X, Greenbaum D, Karro J, Gerstein M . TopNet: a tool for comparing biological sub-networks, correlating protein properties with topological statistics. Nucleic Acids Res 2004; 32:328–337.
Wernicke S, Rasche F . FANMOD: a tool for fast network motif detection. Bioinformatics 2006; 22:1152–1153.
Ramsey S, Orrell D, Bolouri H . Dizzy: stochastic simulation of large-scale genetic regulatory networks (supplementary material). J Bioinform Comput Biol 2005; 3:437–454.
Adalsteinsson D, McMillen D, Elston TC . Biochemical Network Stochastic Simulator (BioNetS): software for stochastic modeling of biochemical networks. BMC Bioinformatics 2004; 5:24.
Aho T, Smolander OP, Niemi J, Yli-Harja O . RMBNToolbox: random models for biochemical networks. BMC Syst Biol 2007; 1:22.
Alves R, Antunes F, Salvador A . Tools for kinetic modeling of biochemical networks. Nat Biotechnol 2006; 24:667–672.
Acknowledgements
I thank Dr Michael Cusick of Dana-Farber Cancer Institute for thoroughly editing the manuscript, Huiling Xue for generating Supplementary information, Table S1, Kai Xia for comments on network analysis tools, Dr Tao Jiang of UC Riverside and Dr Nicholas Baker of Albert Einstein College of Medicine for critical reading of the manuscript and the three anonymous reviewers for their valuable comments. I acknowledge supports by grants from the China National Science Foundation (Grant # 30588001 and 30620120433), National Basic Research Program of China (Grant # 2006CB910700) and funds from the Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Rights and permissions
About this article
Cite this article
Han, JD. Understanding biological functions through molecular networks. Cell Res 18, 224–237 (2008). https://doi.org/10.1038/cr.2008.16
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2008.16
Keywords
This article is cited by
-
Emerging landscape of molecular interaction networks: Opportunities, challenges and prospects
Journal of Biosciences (2022)
-
The landscape of aging
Science China Life Sciences (2022)
-
Protein residue network analysis reveals fundamental properties of the human coagulation factor VIII
Scientific Reports (2021)
-
Differential network analysis of bovine muscle reveals changes in gene coexpression patterns in response to changes in maternal nutrition
BMC Genomics (2020)
-
A biochemical network modeling of a whole-cell
Scientific Reports (2020)
