
Abstract
The Pierre Robin Sequence (PRS), consisting of cleft palate, glossoptosis and micrognathia, is a common human birth defect. However, how this abnormality occurs remains largely unknown. Here we report that neural crest cell (NCC)-specific knockout of transferrin receptor (Tfrc), a well known transferrin transporter protein, caused micrognathia, cleft palate, severe respiratory distress and inability to suckle in mice, which highly resemble human PRS. Histological and anatomical analysis revealed that the cleft palate is due to the failure of palatal shelves elevation that resulted from a retarded extension of Meckel’s cartilage. Interestingly, Tfrc deletion dramatically suppressed both transforming growth factor-β (TGF-β) and bone morphogenetic protein (BMP) signaling in cranial NCCs-derived mandibular tissues, suggesting that Tfrc may act as a facilitator of these two signaling pathways during craniofacial morphogenesis. Together, our study uncovers an unknown function of Tfrc in craniofacial development and provides novel insight into the etiology of PRS.
Similar content being viewed by others


Main
Neural crest cells (NCCs) are highly pluripotent stem cell populations that arise from the neural folds and migrate extensively to different regions, where they differentiate into a broad range of cell types to build up various structures. The cranial NCCs (CNCCs) originate from posterior forebrain and posterior hindbrain and contribute to much of bones, cartilages, cranial nerves, muscles and connective tissues in face and neck.1, 2, 3, 4 During vertebrate craniofacial development, the specification, migration, proliferation and differentiation of CNCCs are tightly controlled by a spatial and temporal signaling network.5 Aberrations in any node of these signaling networks could result in craniofacial malformations, such as cleft lip, cleft palate and mandibular hypoplasia.
The Pierre Robin Sequence (PRS) is a human craniofacial malformation (OMIM: 261800) characterized by U-shaped cleft palate, glossoptosis (backward displacement of the tongue) and micrognathia (abnormally small mandible).6, 7, 8 PRS occurs with an incidence of 1 : 8500 to 1 : 14 000 in live births and results in severe respiratory distress and feeding difficulties in infants.6, 9, 10 Mutations in several genes, including BMP2, SATB2, COL2A1, COL11A1, COL11A2, SOX9 and KCNJ2, have been identified in PRS.11, 12, 13, 14, 15 Mandibular growth retardation is considered to be the most possible cause for PRS-cleft palate.7 In mammals, Meckel’s cartilage has a close relation to the mandibular development. During embryonic development, the extension of Meckel’s cartilage within the mandibular prominence promotes the elongation of mandible, which subsequently assists the flattening of tongue and permits the elevation and followed fusion of palatal shelves.7, 16 Multiple genetic evidences have demonstrated that defective mandible and Meckel’s cartilage development could lead to cleft palate.7, 17, 18, 19, 20, 21, 22
Transferrin receptor (Tfrc) is a homodimeric trans-membrane glycoprotein that binds to iron-bound transferrin and mediates the extracellular iron to be delivered into cells via clathrin-dependent endocytosis.23, 24 Although Tfrc is widely believed to be important for iron acquisition by all mammalian cells, its physiological function for mammal’s development is poorly understood. Here, we report that Tfrc is necessary for embryonic craniofacial development. Mice lacking Tfrc in NCCs exhibit micrognathia, cleft palate, severe respiratory distress and inability to suckle, which highly resemble human PRS. Histological and anatomical analysis revealed that cleft palate results from the failure of palatal shelves elevation, furthermore the failed palatal shelves elevation is due to retarded extension of Meckel’s cartilage that is essential for mandible elongation. Molecularly, the ablation of Tfrc in NCCs significantly suppressed transforming growth factor-β (TGF-β) and BMP signaling in NCCs-derived mandibular tissues. Based on these data, we propose that Tfrc is a facilitator of TGF-β and BMP signaling and Tfrc-mediated activation of these two signaling pathways is essential for craniofacial morphogenesis.
Results
Tfrc deletion in NCCs causes craniofacial defects
Although Tfrc has been studied extensively, the potential role of Tfrc in regulating NCCs development remains unclear. Therefore, we first examined the expression pattern of Tfrc during NCCs development by immunofluorescent staining. Sections from embryonic day 9.5 (E9.5) wild-type embryos were labeled with Tfrc and transcription factor activator protein 2α (AP-2α) antibodies, a marker for premigratory and migratory NCCs.25, 26 Tfrc was broadly and highly expressed in regions populated by cells of neural crest origin, including brachial arch (BA1), frontonasal prominence (FNP) and dorsal root ganglia (Figure 1a). In these regions, Tfrc showed a clustered expression pattern in cytoplasm (Figure 1b).

Conditional deletion of Tfrc in NCCs leads to mandible hypoplasia. (a) Expression pattern of Tfrc in NCCs-derived cells is detected by immunofluorescent staining of Tfrc (red) and AP-2α (green) in E9.5 embryo sections. (b) Magnification of the arrowheads indicated regions in a. Arrows mark the cluster of Tfrc in cell cytoplasm. (c) Expression of Tfrc protein in NCCs-derived palate, maxillary, mandible and skull. Tfrcs were reduced in Wnt1cre;Tfrcf/f mutants compared with littermate controls. (d) Newborn Wnt1cre;Tfrcf/f mutant exhibits shortened mandible (arrow) and no milk in stomach (arrowhead). (e) The ventral view of P0 Wnt1cre;Tfrcf/fmutant shows evident hypoplastic mandible (dotted line) and incomplete oral closure (asterisk). (f, g) P0 Wnt1cre;Tfrcf/f mutants show abundant flatus in intestine (F, arrows) and stomach (g, asterisk). BA1, first branchial arch; BA2, second branchial arch; FNP, frontonasal prominence. Scale bar, 100μm (a), 10μm (b), 5 mm (d), 1.5 mm (e), 2.5 mm (f), 0.83 mm (g)
Besides its expression in NCCs-derived cells, Tfrc is expressed broadly during development, therefore it is not surprising that mice deficient for the Tfrc gene die before E12.5.27 To circumvent this early lethality and investigate the function of Tfrc in NCCs, we generated NCCs-specific Tfrc knockout (KO) mice by firstly crossbreeding Wnt1cre mice28 with homozygous floxed Tfrc mice (Tfrcflox/flox)29 to get Wnt1cre;Tfrcf/+ heterozygote, then crossbreeding Wnt1cre;Tfrcf/+ heterozygote with Tfrcflox/flox to get Wnt1cre;Tfrcf/f mutants. The specificity and efficiency of Tfrc KO were examined by immunoblotting. As compared with control littermates, Wnt1-cre-mediated KO of Tfrc dramatically reduced Tfrc protein level in NCCs-derived craniofacial tissues, including palate, maxillary, mandible and skull (Figure 1c). Wnt1cre;Tfrcf/f mutants were born at expected Mendelian ratios but all died within 24 h after birth. Morphologically, newborn Wnt1cre;Tfrcf/f mutants exhibited micrognathia (Figures 1d and e), incomplete oral closure (Figure 1e), severe respiratory distress (Supplementary Material and Supplementary Movie 1) and inability to suckle, highly resembling the human PRS. Due to these severe defects, the mutant pups sucked air rather than milk into their gastrointestinal tract (Figures 1f and g). The morphologies of other tissues derived from NCCs were also examined in postnatal day 0 (P0) mutant pups. As shown, Tfrc KO in NCCs did not induce abnormalities in these tissues, including craniofacial nerves (Supplementary Figures S1A and B), dorsal root ganglions (Supplementary Figures S1 C–F) and cardiac development (Supplementary Figures S1 G–J). All these results suggest that Tfrc expression in NCCs is required for craniofacial development.
Wnt1cre;Tfrcf/f mutants exhibit cleft palate
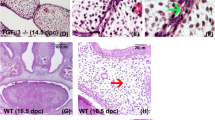
Since phenotypes of respiratory distress and gastrointestinal tract filled with air bubbles are commonly seen in newborn mice with cleft palate, we removed mandible and observed a complete cleft palate in Wnt1cre;Tfrcf/f mutant when compared with control littermate (Figures 2a and b). To determine the onset of cleft palate in mutants, the developments of palate at various embryonic stages were examined by histological staining. In E13.5 controls, the palatal shelves grew vertically down to the two sides of tongue (Figure 2c); then at E14.5, they rotated to a horizontal position above tongue and elongated to fuse with remnant medial edge epithelium (MEE) (Figure 2e), finally the E16.5 and P0 controls exhibited completely fused palate with flat tongues (Figures 2g and i). However, this process was disrupted by Tfrc deletion. The palatal shelves in E13.5 Wnt1cre;Tfrcf/f mutants were well-developed and properly extended to the two sides of tongue (Figure 2d), but they failed to elevate above the tongue (Figure 2f), and consequently the E16.5 and P0 mutants displayed cleft palates with arched tongues (Figures 2h and j). The normal initiation of palatal bone were formed by mesenchymal cells both in Wnt1cre;Tfrcf/f mutants and controls (Figures 2e and f), suggesting that Tfrc is not essential for palatal bone initiation but palatal fusion. Next, to examine the ability to fuse for palatal shelves in vitro, we performed the palatal shelves culture assay. In brief, palatal shelves were dissected from E13.5 embryos and placed in pairs on Millipore filters (EMD Millipore, Merck, KGaA, Darmstadt, Germany) with correct anterior–posterior orientation. By 3 days in culture, all these palatal shelves from Wnt1cre;Tfrcf/f mutants and control littermates fused completely with normal disappearance of MEE (Figures 2k and l), suggesting that palatal shelves in Wnt1cre;Tfrcf/f mutants retain the ability to fuse. In addition, formation of osteoid-like structures were observed in both control and mutant palatal mesenchyme (Figures 2k and l), indicating that the palatal bone initiation was not Tfrc dependent. Together, these results suggest that Tfrc depletion in NCCs leads to failed elevation of palatal shelves and thereby causes cleft palate.

Conditional deletion of Tfrc in NCCs leads to cleft palate. (a and b) The ventral view of oral cavity shows fused palate in P0 control (a), but cleft palate in Wnt1cre;Tfrcf/f mutant (b, arrows). (c and d) The palatal shelves in E13.5 Wnt1cre;Tfrcf/f mutant and control both grow vertically at the two sides of tongue. (e and f) The palatal shelves in E14.5 control mouse have rotated horizontally above tongue and fused with remaining MEE (e, arrow), however, they fail to elevate and remain at the two sides of tongue in Wnt1cre;Tfrcf/f mutant (f). Arrowheads in e and f mark cell aggregates formed normally in Wnt1cre;Tfrcf/f mutant and control, which symbolize the precursors of palatal bone. (g–j) The palatal shelves in E16.5 and P0 controls have completely fused with disappearance of midline epithelium (g and i), in contrast to the cleft palates with arched tongues in Wnt1cre;Tfrcf/f mutants (h and j). (k and l) In vitro organ culture shows palatal shelves in controls (n=17) and Wnt1cre;Tfrcf/f mutants (n=7) all fused with complete disappearance of midline epithelium (asterisk), arrows indicate osteoid-like cell aggregated mass. P, palate; T, tongue; Mc, Meckel’s cartilage. Scale bar, 1.5 mm (a and b), 0.1 mm (c–l)
Craniofacial skeletons are malformed in Wnt1cre;Tfrcf/f mutants
To examine the morphologies of craniofacial skeletons, whole-mount pups of Wnt1cre;Tfrcf/f mutants and control littermates were stained with Alcian blue and Alizarin red to reveal cartilages and bones, respectively. In a line with the observed micrognathia in newborn mutant pups, the mandibular bones in Wnt1cre;Tfrcf/f mutant were significantly shorter than that in control (Figures 3a and b). The secondary cartilage of angular process was nearly missing, while the mandibular condyle and coronoid process were also severely blocked/delayed by Tfrc deletion (Figures 3c and d). Moreover, the mandibular bones in Wnt1cre;Tfrcf/f mutant were excessively curved, which consequently widened the distance between bilateral mandibular bones (Figures 3e and f). The hyoid bone (Figure 3h), tympanic ring and gonial bone (Figure 3j) were also deformed in Wnt1cre;Tfrcf/f mutant when compared with that in control (Figures 3g and i). The cartilages of nasal capsule and gross morphologies of maxillary bones in Wnt1cre;Tfrcf/f mutant and control were comparable (Figures 3k and l). The palatal process of palatine (pppl) and palatal process of maxilla (ppmx) extended horizontally and eventually fused to form the secondary palate (Figures 3k and m) in control. In contrast, these processes for secondary palate formation were disrupted in Wnt1cre;Tfrcf/f mutant, both pppl and ppmx failed to elevate or fuse (Figures 3l and n).

Wnt1cre;Tfrcf/f mutants exhibit craniofacial skeleton defects. (a and b) Alizarin red and Alcian blue staining of bone and cartilage in P0 Wnt1cre;Tfrcf/f mutant and littermate control. Asterisks and arrowheads indicate mandible and tympanic ring, respectively. (c and d) The lateral view of mandible dissected from neonates skeleton preparations. The extension of Meckel’s cartilage is absent, while the distal symphysis formed normally (double arrowheads in d), but smaller size of mandibular bone (arrowheads in d) in Wnt1cre;Tfrcf/f mutant. (e and f) The top view of mandible dissected from neonates skeleton preparations. Note the mis-angled mandibular bone (white line) and widened distance between mandibular bones (two-way arrow) in Wnt1cre;Tfrcf/f mutant. (g and h) The ventral view of hyoid bone and laryngeal cartilages dissected from neonates skeleton preparations. Arrowheads in g indicate articulation between lesser horn and greater horn of hyoid bone in control, but lesser horn and hyoid bone are malformed in Wnt1cre;Tfrcf/f mutant (h). (i and j) The lateral view of tympanic ring dissected from neonates skeleton preparations. Note the shortened tympanic ring and hypoplastic gonial bone (arrow in j) in Wnt1cre;Tfrcf/f mutant. (k and l) The ventral view of skull from neonates skeleton preparations. Note the fusion of bilateral palatal bones in control (dotted line in k), but cleft in Wnt1cre;Tfrcf/f mutant (arrowheads and dotted lines in l). (m and n) The horizontal view of palatal bones isolated from neonates skeleton preparations. Note the fusion of palatal bones in control (asterisk in m), but cleft with remnant of palatal bones in Wnt1cre;Tfrcf/f mutant (arrows in n). (o and p) The lateral view of middle ear cartilages and tympanic ring dissected from neonates skeleton preparations. Note the Meckel’s cartilage extends from distal to proximal otic capsule in control (arrow in o) but not in Wnt1cre;Tfrcf/f mutant (asterisk in p). (q and r) Alcian blue staining of cartilage in E14.5 control and Wnt1cre;Tfrcf/f mutant. Arrow in (q) indicates the proximal arms of Meckel’s cartilage articulate normally with middle ear capsule in control, arrowhead in (r) indicates the failed extension of Meckel’s cartilage to middle ear capsule in Wnt1cre;Tfrcf/f mutant. Note the reduced extension of Meckel’s cartilage in Wnt1cre;Tfrcf/f mutant. (s and t) The ventral view of Meckel’s cartilage in E14.5 embryos. Arrow in s indicates the fusion of Meckel’s cartilages at distal tip, arrowheads in (t) indicate a gap between Meckel’s cartilages in Wnt1cre;Tfrcf/f mutant. Scale bar, 1mm (a–f, q and r), 3 mm (g–j), 1.5 mm (k and l), 4.5 mm (m and n), 2 mm (o and p, s and t). ang.sc, secondary cartilage of the angular process of mandible; cond, condylar process of the mandible; cor, coronoid process of the mandible; gh, greater horn of hyoid; h, hyoid; lh, lesser horn of hyoid; Mec.ae, anterior extremity of the Meckel’s cartilage; nc, nasal capsule; ns, nasal septum; pmx, premaxilla; ppmx, palatal process of maxilla; pppl, palatal process of palatine; pppx, palatal process of premaxilla; th, thyroid cartilage
NCCs-derived Meckel’s cartilage is a significant determinant for mandibular development due to its inductive competence of initiating and regulating the ossification of mandible.30 To test whether the mandible defect was caused by abnormal development of Meckel’s cartilage, we carefully analyzed the effect of Tfrc KO on developmental process of Meckel’s cartilage. In contrast to that Meckel’s cartilages were well-developed and extended from the symphysis of mandibular bones to malleus in P0 control (Figure 3o), no mature Meckel’s cartilages were observed at the proximal end junction to malleus in Wnt1cre;Tfrcf/f mutant (Figure 3p). Further examination showed that the defects of Meckel’s cartilage occur as early as in E14.5 Tfrc KO embryos (Figures 3q–t). Alcian blue staining showed the well-developed Meckel’s cartilage in control embryo (Figure 3q). In contrast, Meckel’s cartilages were obviously smaller and the proximal arms of Meckel’s cartilage were not articulated with middle ear capsule in Wnt1cre;Tfrcf/f mutant (Figure 3r). Moreover, the Meckel’s cartilages in mutant were not fused at the distal tip when compared with that in control (Figures 3s and t). These results indicate that Tfrc is essential for Meckel’s cartilage maturation during embryonic development. Presumably, loss of Tfrc in NCCs primarily cause less developed Meckel’s cartilage, consequently leading to mandible defect. Craniofacial skeletal abnormalities in Wnt1cre;Tfrcf/f mutants were summarized in Supplementary Material Supplementary Table S1.
Cleft palate in Wnt1cre;Tfrcf/f mutant is secondary to mandibular defects
It has been documented that normal mandibular development is required for palate formation. Extension of Meckel’s cartilage promotes the forward growth and elongation of lower jaw, which allows the reposition of tongue from its arched pattern to a flatten position in oral cavity and permits the subsequent palatal shelves elevation and fusion.7, 16 Given that the cleft palate phenotype in Wnt1cre;Tfrcf/f mutant was tightly accompanied by shortened mandible and less matured Meckel’s cartilage, we wondered whether the cleft palate is a secondary effect resulting from mandibular defects. To address this question, we employed Nestin-cre mediated Tfrc KO mice (Nestincre;Tfrcf/f). Wnt1-cre-driven deletion of Tfrc occurs both in mandible and palatal mesenchyme, Nestin-cre-mediated Tfrc ablation happens in the mesenchyme and epithelium of palatal shelves but not in mandible.31 We observed that the newborn Nestincre;Tfrcf/f mutant does not phenocopy the Wnt1cre;Tfrcf/f mutant but displays normal palatogenesis (Supplementary Figure S2). This result suggests that cleft palate in Wnt1cre;Tfrcf/f mutant is not caused by intrinsic defects within palate but most probably is a secondary defect resulting from mandibular defects.
NCC-specific deletion of Tfrc does not affect craniofacial NCCs migration or proliferation
To explore the possible cellular mechanism by which Tfrc controls mandibular development, we first examine whether Tfrc regulates CNCCs migration. Whole-mount AP-2α staining in E9.5 and E10.5 embryos was performed to track the migratory CNCCs, and the results showed comparable expression level and distribution pattern of AP-2α in Wnt1cre;Tfrcf/f mutants and control littermates (Figures 4a and b). To confirm this result, Wnt1cre;Tfrcf/+ mice were crossed with Tfrcf/f;Rosa26-reporter mice to get the Wnt1cre;Tfrcf/f;R26R mutants. As expected, Wnt1cre;Tfrcf/f;R26R mutants also showed mandibular hypoplasia and cleft palate (data not shown). Whole-mount X-gal staining revealed normal CNCCs migration and distribution to FNP, BA1 and BA2 in Wnt1cre;Tfrcf/f;R26R mutants as compared with controls (Figures 4c and d). Next, we examined whether cell proliferation was affected by the loss of Tfrc. The NCC-derived mesenchymal cell proliferation rate within mandibular regions, as measured by BrdU (5-bromo-2′-deoxy-uridine) incorporation, was similar between E13.5 Wnt1-cre; Tfrcf/f mutants and controls (Figures 4e and f). Therefore, these results indicate that Tfrc ablation in NCCs does not affect CNCCs migration or proliferation.

Conditional deletion of Tfrc in NCCs does not affect neural crest migration or cell proliferation. (a and b) Whole-mount AP-2α staining shows migratory neural crest cells in FNP (asterisk), BA1 (arrow) and BA2 (arrowhead). (c and d) Whole-mount X-gal staining shows neural crest cells in FNP (asterisk), BA1 (arrow) and BA2 (arrowhead). (e and f) Immunofluorescent labeling (e) and quantification (f) of BrdU-positive cells in mandibular regions from coronal head sections. At least twenty-five sections were randomly selected from four pairs of E13.5 Wnt1cre;Tfrcf/f mutants and controls. NS, no significant difference. Scale bar, 0.5 mm (a–d), 0.1 mm (e). BA1, first branchial arch; BA2, second branchial arch; FNP, frontonasal prominence
Tfrc is required for osteochondrogenic differentiation
To investigate whether the mandibular cartilage and bone defects in Wnt1cre;Tfrcf/f mutants were due to abnormal osteochondrogenic differentiation, we analyzed the expression of marker genes that are required for osteochondrogenic differentiation in E13.5 mandibular tissues by RT-qPCR. The mRNA levels of chondrocyte genes Sox9 and Col2a1, and osteoblast gene ALP and osteocalcin were all decreased in Wnt1cre;Tfrcf/f mutants (Figure 5a), suggesting an osteochondrogenesis defect in Wnt1cre;Tfrcf/f mandible. To examine whether Tfrc directly regulates osteochondrogenic differentiation, in vitro osteochondrogenic differentiation assay was performed using mesenchymal cells isolated from control and Tfrc mutant mandible. On induction of differentiation in medium with β-glycerophosphate and ascorbic acid, control cells showed time-dependent cartilage matrix depositions and mineralized matrix depositions (Figures 5c–f). In contrast, these depositions were reduced in Tfrc KO cells (Figures 5c–f), suggesting a requirement of Tfrc for osteochondrogenic differentiation. Moreover, expression of Sox9, Col2a1 and ALP was decreased in Tfrc KO cells (Figure 5b), which further supports for a differentiation defect induced by Tfrc deletion. Therefore, these results indicate that Tfrc is required for chondrogenic and osteogenic cell differentiation.

Impaired osteochondrogenic differentiation in Wnt1cre;Tfrcf/f mutants. (a) Expression levels of chondrocyte and osteoblast genes in mandible tissue isolated from E13.5 Wnt1cre;Tfrcf/f mutants and controls. Data shown are normalized ratio of Wnt1cre;Tfrcf/f /Control (mean±S.E.M); student’s t-test; *P<0.05; **P<0.01; ***P<0.001; n≥3. (b) Expression levels of chondrocyte and osteoblast genes in cultured primary mandible mesenchymal cells. Mandible mesenchymal cells were cultured and induced to undergo osteochondrogenic differentiation then collected samples at day 7. Data shown are normalized ratio of Wnt1cre;Tfrcf/f/Control (mean±S.E.M); student’s t-test; **P<0.01; ***P<0.001; n=5. (c–f) Alcian blue (c) and Alizarin red (e) staining show reduced in vitro osteochondrogenic differentiation in Tfrc KO cells. Mandible mesenchymal cells were cultured and induced to undergo osteochondrogenic differentiation, then collected and stained with Alcian blue at day 0, 4, 7 and 14. (d) and (f) quantification of OD 620 nm and OD 450 nm absorbance data from (c and e), respectively. Data shown are mean±S.E.M; student’s t-test; ***P<0.001; control, n=23; Wnt1cre;Tfrcf/f, n=14
Tfrc deletion suppressed TGF-β and BMP signaling in mandible
A couple of signaling pathways including Wnt/β-catenin, Sonic hedgehog (Shh), endothelin, fibroblast growth factor (FGF) family and TGF-β superfamily have been implicated as critical regulators for craniofacial development. We then tested whether Tfrc regulates craniofacial development through these signaling pathways. Axin2, Ptch1, Dlx1, Gsc and Runx2, which are, respectively, typical downstream target of the indicated five pathways, were selected for expression analysis by using RT-qPCR. As shown, only Runx2, a target gene of TGF-β superfamily signaling, was downregulated in Wnt1cre;Tfrcf/f mutants (Figure 6a). Protein expression levels of TGF-βR2, BMPR2 and Runx2 were all decreased in Wnt1cre;Tfrcf/f mutants mandibular tissue; moreover, phosphorylation of smad2 and smad5 (P-samd5 and P-smad2), which, respectively, represents the activation of TGF-β and BMP signaling, were also hugely reduced (Figures 6b and c), suggesting that Tfrc is required for the activation of TGF-β and BMP signaling. Next, we confirmed this result in cultured mandibular mesenchymal cells. Results showed that Tfrc deletion dramatically suppressed the phosphorylation of smad5 and smad2 induced by TGF-β and BMP2 treatment (Figures 6d and e). Combined, these results suggest that Tfrc is critical for the activation of TGF-β and BMP signaling, and Tfrc-dependent activation of these signaling pathways likely contributes to craniofacial morphogenesis by controlling mandibular development.

Loss of Tfrc in NCCs suppresses the activation of TGF-β and BMP signaling. (a) Expression levels of indicated signaling pathways targeted genes in mandible tissues dissected from E13.5 Wnt1cre;Tfrcf/f mutants and controls. Data shown are normalized ratio of Wnt1cre;Tfrcf/f /Control (mean±S.E.M); student’s t-test; NS, no significant difference; ***P<0.001; n≥3. (b) Expression levels of proteins involved in TGF-β and BMP signaling in E13.5 mandible tissues. Data shown are normalized ratio of Wnt1cre;Tfrcf/f /Control (mean±S.E.M); student’s t-test; *P<0.05; **P<0.01; n≥3. (c) Immunostaining of P-Smad5 in E13.5 mandible sections. P, palate; T, tongue; Mc, Meckel’s cartilage. Scale bar, 100 μm. (d and e) Immunoblotting and quantification of P-smad5 (c) and P-smad2 (d) in cultured primary mandibular mesenchymal cells. Mandibular mesenchymal cells were cultured and induced to undergo osteochondrogenic differentiation for 7 days, then treated with BMP2 (100 ng/ml) or Tgfβ3 (50 ng/ml) for 30 min at 37 °C. Data shown are normalized ratio of KO/Control (mean±S.E.M); student’s t-test; *P<0.05; **P<0.01; n=3
Discussion
In this study, we investigated the function of Tfrc in NCCs by using a Wnt1cre-LoxP system and found a previously unknown role of Tfrc during craniofacial development. Specific deletion of Tfrc in NCCs leads to craniofacial defects including cleft palate, severe respiratory distress and inability to suckle, resembling the human PRS (OMIM 261800). Consequently, Wnt1cre;Tfrcf/f mutant pups die within 24 h after birth. Further examinations indicate that mandible hypoplasia resulted from Meckel’s cartilage development defects is presumably a primary cause for cleft palate in Wnt1cre;Tfrcf/f mutants. Thus, our study provides evidence that Tfrc-mediated signaling in NCCs is crucial for craniofacial development.
Wnt1cre;Tfrcf/f mutant mice simultaneously exhibit the cleft palate and hypoplastic mandible phenotypes. Timing histological and anatomical analysis revealed that the cleft palate in Wnt1cre;Tfrcf/f mutants is directly caused by failed elevation of palatal shelves, while hypoplastic mandible resulted from retarded growth of Meckel’s cartilage. However, the lack of either cleft palate or hypoplastic mandible in Nestin-Cre-mediated Tfrc KO mice suggests a different mechanism. Considering that Wnt1-cre-driven deletion of Tfrc occurs in mandible and palatal mesenchyme, while Nestin-cre-mediated Tfrc ablation mainly happens in the mesenchyme and epithelium of palatal shelves but not in mandible, we propose that cleft palate in Wnt1cre;Tfrcf/f mutant is a second consequence due to mandibular development defects. Mechanistically, Tfrc KO in NCCs primarily reduce the extension of Meckel’s cartilage that is required for correct forward growth and elongation of lower jaw, then consequently cause mandibular hypoplasia. As a second result, defective mandibular maturation restricts the elevation of palatal shelves from vertical to horizontal position and then leads to cleft palate in Wnt1cre;Tfrcf/f mutants. Our finding is consistent to previous reports that obstruction of mandibular growth leads to cleft palate in several mutants, such as Hoxa2−/−,17 Egfr−/−,18 Dmm,7 Wnt1Cre;Alk2flox/flox mutants,19 Snai1+/–;Snai2−/−,20 csp1, Prdm16Gt683Lex mutants 21 and Ptprs−/−;Ptprf−/−.22 Interestingly, cleft palate in human PRS is also due to the failure of tongue translocation or mandibular growth defects, suggesting that Wnt1cre;Tfrcf/f mutant mice not only morphologically but also mechanistically resemble human PRS.
What is the cellular mechanism for mandibular defects in Wnt1cre;Tfrcf/f mutant? Fine control of NCC migration, proliferation and differentiation is essential for Meckel’s cartilage processing and mandible elongation. Our analysis revealed that Tfrc is not required for NCCs migration or proliferation. However, the differentiation of chondrogenic and osteogenic cell within mandible are severely suppressed by Tfrc deletion (Figure 5). Further molecular analysis showed that the levels of Sox9, Col2a1, ALP and Osteocalcin, which are highly expressed within differentiated cells, are decreased in Tfrc KO cells (Figures 5a and b), confirming that the differentiation process is delayed by Tfrc ablation. Thus, these results indicate that the hypoplastic mandible in Wnt1cre;Tfrcf/f mutant is likely a result of impaired chondrogenic and osteogenic differentiation.
What is the molecular mechanism for mandibular defects in Wnt1cre;Tfrcf/f mutant? TGF-β superfamily signaling is known to perform essential roles in mandible and Meckel’s cartilage development. Disruption of TGF-β superfamily signaling by either complete or conditional inactivation of Bmp4, TGF-β2, TGF-βR2, Alk5, Alk2 or Smad4 results in abnormal development of Meckel’s cartilage and mandible, which lead to cleft palate.19, 32, 33, 34, 35, 36, 37 In the present study, we found that Tfrc deletion in NCCs dramatically suppresses both TGF-β and BMP signaling. In consistence, Tfrc KO mice show cleft palate and hypoplastic mandible that are similar to the phenotypes observed in mice lacking TGF-β or BMP signaling. Our data thus suggest that Tfrc is a critical regulator for TGF-β superfamily signaling activation during craniofacial development.
How does Tfrc control TGF-β superfamily signaling? Here, we speculate that Tfrc may control the endocytosis of TGF-β and BMP receptors and is then required for TGF-β and BMP activation. First, TGF-β and BMP signaling activation is dependent on clathrin-mediated endocytosis of TGF-β and BMP receptors.38, 39, 40 Second, adaptor protein 2 (AP-2), an essential adaptor for clathrin-mediated endocytosis, is identified to interact with TGF-β and BMP receptors.41, 42 Third, local clustering of Tfrc at cell surface promotes clathrin-coated pits initiation and clathrin lattices formation,43, 44, 45 and a tight association between AP-2 and Tfrc was detected in our study (data not shown) and in previous literature.46 Fourth, Tfrc deletion in NCCs suppresses both TGF-β and BMP signaling. So, it is plausible that Tfrc functions as a facilitator to control TGF-β and BMP receptor endocytosis via binding clathrin adaptor AP-2. Of note, Tfrc deletion may also affect caveolin-dependent endocytosis of TGF-β and BMP receptors. It has been documented that TGF-β and BMP receptors are internalized into cytoplasm through two distinct endocytic pathways, clathrin-mediated endocytosis and caveolin-dependent endocytosis. Clathrin-mediated endocytosis is important for the signaling activation, whereas caveolin-dependent endocytosis is required for receptor degradation.38, 39, 40 We indeed found that Tfrc deletion strikingly decreased the protein level of both TGF-β and BMP receptors, indicating increased degradation of TGF-β and BMP receptors in response to Tfrc deletion. We hypothesize that this effect is possibly due to accelerated caveolin-dependent endocytosis of TGF-β and BMP receptors in Tfrc KO cells. Likely, Tfrc deletion switches the internalization pathway of TGF-β and BMP receptors from clathrin-mediated to caveolin-mediated endocytosis and thereby leads more receptors to be destined for degradation. Certainly, it is also possible that Tfrc negatively regulates caveolin-dependent endocytosis of TGF-β and BMP receptors in a direct manner. Further experiments will be needed to investigate these possible mechanisms in detail.
Materials and Methods
Generation of Wnt1cre;Tfrcf/f mutant mice and genotyping
All mice were kept in specific pathogen-free standard condition and all experiments were conducted in accordance with the guidelines and under the approval of Animal Care Committees at Sichuan University. The generation of Wnt1cre;Tfrcf/f mutants were obtained by cross-mating of Tfrcflox/flox mice (from Andrews)29 with heterozygote Wnt1cre; Tfrcf/+. Wnt1cre; Tfrcf/+heterozygous had no obvious phenotype, control littermates were taken as Tfrcf/+, Tfrcf/f or Wnt1cre; Tfrcf/+. Genotyping was performed by PCR using primers listed in Supplementary Material and Supplementary Table S2.
Skeletal preparations
Embryonic or neonatal controls and Wnt1cre;Tfrcf/f mutants were de-skinned, eviscerated and fixed in 100% ethanol for 24 h, then followed by 100% acetone to remove fat. Skeletons were stained in Alcian blue solution (150 mg/l Alcian blue, 80% ethanol and 20% acetic acid) for 48 h at 37 °C, then washed in 95% ethanol for 8-12 h and cleared in 2% KOH solution for 24 h, followed by staining overnight in Alizarin red solution (50 mg/l Alizarin red in 2% KOH). Finally, the skeletons were cleared in water and stored in 20% glycerol.
Histological staining
For immunostaining, antibodies against Tfrc and AP-2α were purchased from Invitrogen (136800) and Epitomics (3154-1) and used at 1 : 400.
Whole-mount immunohistochemistry was performed as previously described.47 AP-2α antibody was used at 1 : 100.
Whole-mount X-gal staining was performed using the In Situ β-galactosidase Staining Kit (Beyotime, Haimen, Jiangsu, China, RG0039), following the manufacturer’s recommendations.
For histological examination, embryos and neonatal mice were fixed in 4% paraformaldehyde overnight at 4 °C, then dehydrated in gradient sucrose solution and embedded in OCT, and cryosectioned at 14 μm. Sections were stained with hematoxylin and eosin following standard procedures.
Cell proliferation assay
Cell proliferation was evaluated by DNA synthesis activity through intraperitoneal BrdU injection (50 μg/g body weight) for 1.5 h, then mice were killed and embryos were fixed in 4% paraformaldehyde. Sections were treated with 2 N HCl at 37 °C for 20 min then neutralized with 0.1 M sodium borate for 10 min. BrdU antibody (Santa Cruz, Dallas, TX, USA, sc32323, 1 : 200) was used to detect proliferative cells.
Palatal shelves culture
Palatal shelves culture was performed as previously described with minor modification.33, 48 Briefly, palatal shelves were dissected from E13.5 embryos and placed on Millipore filters as the two segments just touching at the medial edge, cultured for 3 days and fixed in 4% paraformaldehyde followed by histological hematoxylin and eosin analysis.
Immunoblotting analysis
Antibodies used in immunoblotting analysis: Tfrc (Invitrogen, Waltham, MA, USA, 136800), Phospho-Smad5 (Epitomics, Burlingame, CA, USA, 2224-1), Phospho-Smad2 (Cell Signaling Technology, 3101), BMPR2 (Epitomics, S0778), TgfβR2 (Cell Signaling Technology, Burlingame, CA, USA, 3713), Runx2 (Millipore, Merck, KGaA, Darmstadt, Germany, 05-1478), β-actin (Santa Cruz, sc-47778). The immunoblotting analysis followed standard procedures.
Mandibular mesenchymal cell culture
Primary mandibular mesenchymal cell culture and osteochondrogenic differentiation were performed as previously described with minor modification.49, 50 Briefly, mandible from E13.5 embryo was dissected and incubated with 0.25% trypsin at 37 °C for 30 min. Isolated cells were plated at a density of 3 × 104 cells/cm2 and cultured in complete medium (DMEM containing 10% FBS supplemented with penicillin, streptomycin, l-glutamate and sodium pyruvate). After 24 h (day 0), culture medium was supplemented with 10 mM β-glycerophosphate, 50 μg/ml ascorbic acid and 2.5 μM retinoic acid to induce osteochondrogenic differentiation. Followed by indicating days, cells were fixed then stained with Alcian blue and Alizarin red to evaluate cell differentiation, the staining and quantification method were performed as previously described.50, 51
Real-time quantitative PCR
Total RNA was extracted using the RNeasy kit (QIAGEN, Hilden, Germany, 74134), and cDNA was synthesized with the TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (Transgen, Beijing, China, AT311). Quantitative real-time PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems, Waltham, MA, USA, 4367659). Primer sequences used for qPCR were listed in Supplementary Material and Supplementary Table S2, according to previously published results.22, 52 Expression levels of mRNA were normalized to β-actin.
Statistical analysis
Statistical analyses were performed using two-tailed Student’s t-test. Data are presented as mean±S.E.M; P-value<0.05 indicated significant difference.
Abbreviations
- AP-2:
-
adaptor protein 2
- BA:
-
branchial arch
- BMP:
-
bone morphogenetic protein
- BrdU:
-
5-bromo-2′-deoxy-uridine
- CNCCs:
-
cranial neural crest cells
- E:
-
embryonic day
- FNP:
-
frontonasal prominence
- KO:
-
knockout
- MEE:
-
medial edge epithelium
- NCCs:
-
neural crest cells
- P:
-
postnatal day 0
- Ppmx:
-
palatal process of maxilla
- Pppl:
-
palatal process of palatine
- PRS:
-
Pierre Robin Sequence
- Tfrc:
-
transferrin receptor
- TGF-β:
-
transforming growth factor-β
References
Farlie PG, McKeown SJ, Newgreen DF . The neural crest: basic biology and clinical relationships in the craniofacial and enteric nervous systems. Birth Defects Res C Embryo Today 2004; 72: 173–189.
Gammill LS, Bronner-Fraser M . Neural crest specification: migrating into genomics. Nat Rev Neurosci 2003; 4: 795–805.
Knecht AK, Bronner-Fraser M . Induction of the neural crest: a multigene process. Nat Rev Genet 2002; 3: 453–461.
Mayor R, Theveneau E . The neural crest. Development 2013; 140: 2247–2251.
Chai Y, Maxson RE Jr . Recent advances in craniofacial morphogenesis. Dev Dyn 2006; 235: 2353–2375.
Izumi K, Konczal LL, Mitchell AL, Jones MC . Underlying genetic diagnosis of Pierre Robin sequence: retrospective chart review at two children's hospitals and a systematic literature review. J Pediatr 2012; 160: 645–650 e642.
Ricks JE, Ryder VM, Bridgewater LC, Schaalje B, Seegmiller RE . Altered mandibular development precedes the time of palate closure in mice homozygous for disproportionate micromelia: an oral clefting model supporting the Pierre-Robin sequence. Teratology 2002; 65: 116–120.
Robin P . Glossoptosis due to atresia and hypotrophy of the mandible. Am J Dis Child 1934; 48: 541–547.
Printzlau A, Andersen M . Pierre Robin sequence in Denmark: a retrospective population-based epidemiological study. Cleft Palate Craniofac J 2004; 41: 47–52.
Sheffield LJ, Reiss JA, Strohm K, Gilding M . A genetic follow-up study of 64 patients with the Pierre Robin complex. Am J Med Genet 1987; 28: 25–36.
Melkoniemi M, Koillinen H, Mannikko M, Warman ML, Pihlajamaa T, Kaariainen H et al. Collagen XI sequence variations in nonsyndromic cleft palate, Robin sequence and micrognathia. Eur J Hum Genet 2003; 11: 265–270.
Britanova O, Depew MJ, Schwark M, Thomas BL, Miletich I, Sharpe P et al. Satb2 haploinsufficiency phenocopies 2q32-q33 deletions, whereas loss suggests a fundamental role in the coordination of jaw development. Am J Hum Genet 2006; 79: 668–678.
Jakobsen LP, Ullmann R, Christensen SB, Jensen KE, Molsted K, Henriksen KF et al. Pierre Robin sequence may be caused by dysregulation of SOX9 and KCNJ2. J Med Genet 2007; 44: 381–386.
Benko S, Fantes JA, Amiel J, Kleinjan DJ, Thomas S, Ramsay J et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet 2009; 41: 359–364.
Sahoo T, Theisen A, Sanchez-Lara PA, Marble M, Schweitzer DN, Torchia BS et al. Microdeletion 20p12.3 involving BMP2 contributes to syndromic forms of cleft palate. Am J Med Genet A 2011; 155 A: 1646–1653.
Bush JO, Jiang R . Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development 2012; 139: 231–243.
Gendron-Maguire M, Mallo M, Zhang M, Gridley T . Hoxa-2 mutant mice exhibit homeotic transformation of skeletal elements derived from cranial neural crest. Cell 1993; 75: 1317–1331.
Miettinen PJ, Chin JR, Shum L, Slavkin HC, Shuler CF, Derynck R et al. Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nat Genet 1999; 22: 69–73.
Dudas M, Sridurongrit S, Nagy A, Okazaki K, Kaartinen V . Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech Dev 2004; 121: 173–182.
Murray SA, Oram KF, Gridley T . Multiple functions of Snail family genes during palate development in mice. Development 2007; 134: 1789–1797.
Bjork BC, Turbe-Doan A, Prysak M, Herron BJ, Beier DR . Prdm16 is required for normal palatogenesis in mice. Hum Mol Genet 2010; 19: 774–789.
Stewart K, Uetani N, Hendriks W, Tremblay ML, Bouchard M . Inactivation of LAR family phosphatase genes Ptprs and Ptprf causes craniofacial malformations resembling Pierre-Robin sequence. Development 2013; 140: 3413–3422.
Aisen P . Transferrin receptor 1. Int J Biochem Cell Biol 2004; 36: 2137–2143.
Cheng Y, Zak O, Aisen P, Harrison SC, Walz T . Structure of the human transferrin receptor-transferrin complex. Cell 2004; 116: 565–576.
Brewer S, Feng W, Huang J, Sullivan S, Williams T . Wnt1-Cre-mediated deletion of AP-2alpha causes multiple neural crest-related defects. Dev Biol 2004; 267: 135–152.
Mitchell PJ, Timmons PM, Hebert JM, Rigby PW, Tjian R . Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes Dev 1991; 5: 105–119.
Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC . Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet 1999; 21: 396–399.
Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP . Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol 1998; 8: 1323–1326.
Chen AC, Donovan A, Ned-Sykes R, Andrews NC . Noncanonical role of transferrin receptor 1 is essential for intestinal homeostasis. Proc Natl Acad Sci USA 2015; 112: 11714–11719.
Frommer J, Margolies MR . Contribution of Meckel's cartilage to ossification of the mandible in mice. J Dent Res 1971; 50: 1260–1267.
Liu W, Sun X, Braut A, Mishina Y, Behringer RR, Mina M et al. Distinct functions for Bmp signaling in lip and palate fusion in mice. Development 2005; 132: 1453–1461.
Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997; 124: 2659–2670.
Ito Y, Yeo JY, Chytil A, Han J, Bringas P Jr, Nakajima A et al. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development 2003; 130: 5269–5280.
Liu W, Selever J, Murali D, Sun X, Brugger SM, Ma L et al. Threshold-specific requirements for Bmp4 in mandibular development. Dev Biol 2005; 283: 282–293.
Dudas M, Kim J, Li WY, Nagy A, Larsson J, Karlsson S et al. Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev Biol 2006; 296: 298–314.
Ko SO, Chung IH, Xu X, Oka SJ, Zhao H, Cho ES et al. Smad4 is required to regulate the fate of cranial neural crest cells. Dev Biol 2007; 312: 435–447.
Oka K, Oka S, Sasaki T, Ito Y, Bringas P, Nonaka K et al. The role of TGF-beta signaling in regulating chondrogenesis and osteogenesis during mandibular development. Dev Biol 2007; 303: 391–404.
Hartung A, Bitton-Worms K, Rechtman MM, Wenzel V, Boergermann JH, Hassel S et al. Different routes of bone morphogenic protein (BMP) receptor endocytosis influence BMP signaling. Mol Cell Biol 2006; 26: 7791–7805.
Satow R, Kurisaki A, Chan TC, Hamazaki TS, Asashima M . Dullard promotes degradation and dephosphorylation of BMP receptors and is required for neural induction. Dev Cell 2006; 11: 763–774.
Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL . Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol 2003; 5: 410–421.
Yao D, Ehrlich M, Henis YI, Leof EB . Transforming growth factor-beta receptors interact with AP2 by direct binding to beta2 subunit. Mol Biol Cell 2002; 13: 4001–4012.
Bragdon B, Thinakaran S, Bonor J, Underhill TM, Petersen NO, Nohe A . FRET reveals novel protein-receptor interaction of bone morphogenetic proteins receptors and adaptor protein 2 at the cell surface. Biophys J 2009; 97: 1428–1435.
Iacopetta BJ, Rothenberger S, Kuhn LC . A role for the cytoplasmic domain in transferrin receptor sorting and coated pit formation during endocytosis. Cell 1988; 54: 485–489.
Miller K, Shipman M, Trowbridge IS, Hopkins CR . Transferrin receptors promote the formation of clathrin lattices. Cell 1991; 65: 621–632.
Liu AP, Aguet F, Danuser G, Schmid SL . Local clustering of transferrin receptors promotes clathrin-coated pit initiation. J Cell Biol 2010; 191: 1381–1393.
Ohno H, Stewart J, Fournier MC, Bosshart H, Rhee I, Miyatake S et al. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 1995; 269: 1872–1875.
Mark M, Lufkin T, Vonesch JL, Ruberte E, Olivo JC, Dolle P et al. Two rhombomeres are altered in Hoxa-1 mutant mice. Development 1993; 119: 319–338.
Taya Y, O'Kane S, Ferguson MW . Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development 1999; 126: 3869–3879.
Iwata J, Hosokawa R, Sanchez-Lara PA, Urata M, Slavkin H, Chai Y . Transforming growth factor-beta regulates basal transcriptional regulatory machinery to control cell proliferation and differentiation in cranial neural crest-derived osteoprogenitor cells. J Biol Chem 2010; 285: 4975–4982.
Zhou Z, Xie J, Lee D, Liu Y, Jung J, Zhou L et al. Neogenin regulation of BMP-induced canonical Smad signaling and endochondral bone formation. Dev Cell 2010; 19: 90–102.
Tataria M, Quarto N, Longaker MT, Sylvester KG . Absence of the p53 tumor suppressor gene promotes osteogenesis in mesenchymal stem cells. J Pediatr Surg 2006; 41: 624–632 discussion 624-632.
Wu JY, Aarnisalo P, Bastepe M, Sinha P, Fulzele K, Selig MK et al. Gsalpha enhances commitment of mesenchymal progenitors to the osteoblast lineage but restrains osteoblast differentiation in mice. J Clin Invest 2011; 121: 3492–3504.
Acknowledgements
We thank Dr Nancy C Andrews for providing Tfrcflox/flox homozygous mice. This work was partially supported by grants from the Ministry of Science & Technology-China (2014CB964600, 2012CB966800), the National Science Foundation of China (31301125, 31071283) and Shenzhen Key Laboratory for Molecular Biology of Neural Development (ZDSY20120617112838879).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Agostini
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lei, R., Zhang, K., Liu, K. et al. Transferrin receptor facilitates TGF-β and BMP signaling activation to control craniofacial morphogenesis. Cell Death Dis 7, e2282 (2016). https://doi.org/10.1038/cddis.2016.170
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2016.170
This article is cited by
-
Gene regulatory network from cranial neural crest cells to osteoblast differentiation and calvarial bone development
Cellular and Molecular Life Sciences (2022)
-
Investigating the molecular control of deer antler extract on articular cartilage
Journal of Orthopaedic Surgery and Research (2021)
-
The prion protein is embedded in a molecular environment that modulates transforming growth factor β and integrin signaling
Scientific Reports (2018)
-
GWAS reveals loci associated with velopharyngeal dysfunction
Scientific Reports (2018)

{kind=link}
{kind=link}