
Abstract
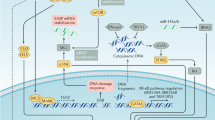
Senescence is characterized by permanent cell-cycle arrest despite continued viability and metabolic activity, in conjunction with the secretion of a complex mixture of extracellular proteins and soluble factors known as the senescence-associated secretory phenotype (SASP). Cellular senescence has been shown to prevent the proliferation of potentially tumorigenic cells, and is thus generally considered a tumor suppressive process. However, some SASP components may act as pro-tumorigenic mediators on premalignant cells in the microenvironment. A limited number of studies indicated that protein kinase C (PKC) has a role in senescence, with different isoforms having opposing effects. It is therefore important to elucidate the functional role of specific PKCs in senescence. Here we show that PKCη, an epithelial specific and anti-apoptotic kinase, promotes senescence induced by oxidative stress and DNA damage. We further demonstrate that PKCη promotes senescence through its ability to upregulate the expression of the cell cycle inhibitors p21Cip1 and p27Kip1 and enhance transcription and secretion of interleukin-6 (IL-6). Moreover, we demonstrate that PKCη creates a positive loop for reinforcing senescence by increasing the transcription of both IL-6 and IL-6 receptor, whereas the expression of IL-8 is specifically suppressed by PKCη. Thus, the presence/absence of PKCη modulates major components of SASP. Furthermore, we show that the human polymorphic variant of PKCη, 374I, that exhibits higher kinase activity in comparison to WT-374V, is also more effective in IL-6 secretion, p21Cip1 expression and the promotion of senescence, further supporting a role for PKCη in senescence. As there is now considerable interest in senescence activation/elimination to control tumor progression, it is first crucial to reveal the molecular regulators of senescence. This will improve our ability to develop new strategies to harness senescence as a potential cancer therapy in the future.
Similar content being viewed by others


Main
Cellular senescence refers to permanent cell-cycle arrest, resulting in stable and long term loss of proliferative capacity, despite continued cell viability and metabolic activity. Senescence was initially identified as the process that limits the replicative life span of cultured human cells, due to the gradual loss of telomeric DNA at the ends of chromosomes (replicative senescence), generating a persistent DNA damage response (DDR).1 However, senescence can also be induced in the absence of detectable telomere loss or dysfunction by various cellular and environmental stressors, such as culture shock, ionizing radiation or prolonged exposure to substantial doses of oxidative stress, generally known as stress-induced premature senescence.2 The definition of senescence was broadened to include oncogene-induced senescence also known as stress or aberrant signaling induced senescence3 Cellular senescence appears to be an anti-proliferative process that limits the progression of damaged cells. Thus, in addition to acting as a potent barrier to tumorigenesis, senescence contributes to the cytotoxicity of certain anti-cancer agents, thereby dictating the outcome of chemotherapy treatment.4, 5 Evidence of the existence of premature senescence in vivo has accumulated, supporting a role for senescence in tumor suppression. For example, naevi on human skin were shown to contain oncogenic mutations, have undergone senescence and therefore failed to develop into malignant tumors.6
Senescent cells have characteristic features, displaying a large and flat morphology, an increase in senescence-associated β-galactosidase (SA-β-gal) activity,7 and in some cell types, a distinct change in chromatin organization known as senescence-associated heterochromatin foci.8 In addition to executing a cell-cycle arrest program, senescent cells undergo massive changes in the expression of genes thought to influence the tissue microenvironment in vivo. Thus, in addition to repressing genes related to proliferation, senescent cells often secrete inflammatory cytokines and immune modulators, downregulate extracellular matrix proteins and upregulate enzymes that degrade extracellular matrix, resulting in the senescence-associated secretory phenotype (SASP) (also called the senescence-messaging secretome9). The senescence secretome has tumor suppressive effects, inducing growth arrest and stimulating the immune system to clear senescent cells. However, components of the secretome can induce pro-tumorigenic effects on premalignant cells in the microenvironment.1 These non-cell-autonomous activities of SASP show the functional relevance of senescence in pathophysiology, in particular in the tumor microenvironment, enhancing tumorigenesis of neighboring cells and playing a role in the decline of organ function with aging.1, 10, 11
Several studies indicated that protein kinase C (PKC) members have a role in senescence,12, 13, 14 sometimes exhibiting opposing effects. It was also suggested that individual PKCs trigger distinct responses when activated in different phases of the cell cycle via a common mechanism that involves the cyclin-dependent kinase (CDK) inhibitor p21Cip1.14 PKCδ was shown to be involved in activating senescence in primary diploid cells.13 Activation of PKCα in lung cancer cells during the S phase of the cell cycle led to G2/M arrest and cellular senescence, an effect that involved p21Cip1 upregulation and irreversible inhibition of cell proliferation. As these lung cancer cells do not express p16INK4a or p53, the upregulation of p21Cip1 was p53 independent.14 While PKCα enhanced senescence, activation of the atypical PKCδ isoform suppressed senescence in breast and glioblastoma cells.12 PKCι depletion led to an increased number of senescent cells, showing no requirement for p53, p16INK4a or ARF, but was markedly dependent on p21Cip1.
Here we show that the novel PKC isoform, PKCη, promotes senescence induced by oxidative stress and DNA damage. The PKCη isoform has a unique tissue distribution and is primarily expressed in epithelial tissues and in cells with high turnover.15 PKCη was found to be involved in diverse cellular functions including terminal differentiation, proliferation and secretion.16, 17 The mechanism of action described in some of these studies involved modulation of cell-cycle components16, 18, 19, 20, 21, 22 and a role in cell cycle progression at the G1 to S phase.16 PKCη was shown to activate a cellular program that includes increased expression of cyclin E, as well as the induced expression of the cyclin-dependent kinase inhibitor p21Cip1.16, 20 Moreover, PKCη formed a complex with Cdk2 and cyclin E in the perinuclear region22 and was shown to phosphorylate p21Cip1.21
Recent studies suggest that PKCη plays a special role in the response to stress and regulation of apoptosis.23, 24, 25 It provides protection against apoptosis induced by chemotherapeutic drugs in Hodgkin's lymphoma cell lines and breast adenocarcinoma MCF-7 cells.26, 27 In addition, PKCη expression was found to correlate with drug resistance and drug-resistance associated genes in patients with breast cancer,28 ovarian cancer29 and AML blasts.30 PKCη expression and membrane localization were shown to be markers for a poor prognosis in lung and breast cancers, respectively.31, 32
Notably, the PKCη gene (PRKCH) was recently identified as a risk factor for cerebral infarction.33 Its gene polymorphism, including a SNP in the coding region (changing valine to isoleucine at position 374 of the kinase domain), was also associated with increased susceptibility for rheumatoid arthritis in Japanese and Chinese populations,34, 35 as well as a risk of severe gastric atrophy.36
Here we show that PKCη promotes senescence induced by oxidative stress and DNA damage via its ability to upregulate the expression of the cell cycle inhibitors p21Cip1 and p27Kip1, and create a positive loop for reinforcing senescence by enhancing the expression of both interleukin-6 (IL-6) and its receptor. Notably, the expression of IL-8, another major component of SASP, was specifically suppressed by PKCη expression, further demonstrating its ability to modulate SASP. Moreover, we show that in comparison to WT-374V, the polymorphic variant 374I of PKCη exhibits enhanced kinase activity, increased secretion of IL-6, enhanced expression of p21Cip1 and higher levels of senescence, further supporting a role for PKCη in senescence.
Results
PKCη expression enhances markers of senescence upon oxidative stress and chemotherapy-induced DNA damage
One of the characteristic markers of senescent cells is an increase in senescence-associated β-galactosidase.7 Oxidative stress and chemotherapy drugs, such as etoposide and doxorubicine were previously shown to induce senescence in MCF-7 cells.37, 38 MCF-7 cells expressing sh-PKCη (clones 2-2, 3-3) and scrambled control cells (clone 5-3) were treated with H2O2 or etoposide under conditions that induce senescence. As seen in Figure 1, in response to H2O2 or etoposide, PKCη-knockdown cells showed significantly and reproducibly lower number of SA-β-galactosidase-stained cells compared with scrambled control cells (Figures 1a and b). Similar results were obtained with doxorubicin (data not shown).

PKCη expression enhances senescence upon oxidative stress and etoposide-induced DNA damage. (a) MCF-7 clones expressing sh-PKCη (sh η 3-3 and sh η 2-2) or scrambled control cells (sh scr 5-3) were treated with 150 μM H2O2 or 400 μM etoposide for 2 h. Fresh medium was added for 96 h. Cells were stained both for Hoechst and SA-β-Gal as described in Materials and Methods. The total cell number was determined by nuclear fluorescent staining. (b) SA-β-gal representative staining of sh-PKCη and scrambled control clones are shown (original magnification × 100 and × 400 for untreated and treated cells, respectively). (c) The untreated control cells and the H2O2-treated senescent cultures of the different clones (sh scr 5-3, sh η 3-3 and sh η 2-2) were replated (1 × 105 cells of each clone) and grown in fresh medium. Cell proliferation was determined using the colony forming assay as described in Materials and Methods. (d) Shown is the quantification of the number of colonies stained by crystal violet shown in c. Results are the average of at least three experiments. Error bars represent the S.D.; two-tailed, unpaired sample t-test statistical analysis is shown: ^ indicates statistical significance compared with untreated cells of the same clone, *indicates statistical significance compared with other clones. *P≤0.05, **P≤0.001, ^^^/***P≤0.0001
Senescence is characterized by permanent irreversible cell-cycle arrest. Therefore, we assessed the number of proliferating cells after the implementation of senescence. As shown in Figures 1c and d a lower number of proliferating cells were regrown out of senescent cultures of scrambled (sh scr 5-3) control cells compared with PKCη-knockdown (sh 3-3 and sh 2-2) cells in colony formation assays. Viability assays (Neutral Red) with additional PKCη-knockdown clones (sh 4-2 and sh 3-5) showed similar results (data not shown). Thus, these results most likely reflect the higher percentage of senescent cells that did not proliferate in PKCη-expressing scrambled (sh scr 5-3) control cells. Several PKCη-knockdown clones were employed in the experiments presented here as these clones lose their silencing abilities over time. Nonetheless, silencing was verified in each experiment for each clone.
The fact that proliferation is blocked in senescence is accompanied by the upregulation of cell cycle control genes. Different studies have indicated that the cell cycle inhibitor p21Cip1 is required to induce senescence. Our results showed increased expression of p21Cip1 and p27Kip1 in PKCη-expressing cells (sh scr 5-3), which was markedly reduced in PKCη-knockdown cells upon etoposide treatment (Figure 2). Cell-cycle arrest upon etoposide treatment was verified by decreased phosphorylation of the retinoblastoma (Rb) protein compared with untreated control cells. In addition, DcR2, another senescence marker,39 was elevated in PKCη expressing cells (Figure 2a).

PKCη-knockdown reduces senescence markers. MCF-7 clones expressing sh-PKCη (sh2-2) or scrambled control cells (scr 5-3) were treated with (a) 400 μM etoposide or (b) 150 μM H2O2 for 2 h. Fresh medium was added for 96 h followed by cell lysis. The senescence markers p21Cip1, p27Kip1, DcR2 and pRB, along with the DDR marker γ−H2AX were detected using Western blot analysis and specific antibodies. β-Actin was used as a marker for equal protein loading. The results shown are representative of three independent experiments. (c) Average percentage of DNA in comet tail. Cells were treated by etoposide as described above, detached by trypsin at the indicated time points, counted and subjected to single-cell electrophoresis as described in Materials and Methods. Results are the average of the volumes of comet tails of at least 250 cells per time point (A minimum of 50 different fields were photographed in three separate experiments). Error bars represent the S.D.; two-tailed, unpaired sample t-test statistical analysis is shown: ^ indicates statistical significance compared with untreated cells of the same clone, * indicates statistical significance compared with other clones. ^P≤0.05 and ^^^/***P≤0.0001. (d) Shown are representative pictures of comet assay analyses of sh-PKCη and scrambled control clones at indicated time points
Etoposide is known to cause DNA damage, triggering DDR, which is one of the mechanisms underlying senescence. γ-H2A histone family, member X (γ-H2AX) phosphorylation at Ser139 is a marker for DNA breaks, initiating DDR. Our results show increased phosphorylation of H2AX in PKCη-knockdown cells under senescence-inducing conditions, triggered by etoposide (Figure 2a) or H2O2 (Figure 2b). The phosphorylation of γ-H2AX may reflect the extent of DNA breaks but also the cellular response to DNA damage. Therefore, to directly evaluate the extent of DNA breaks, we performed comet assays (Figures 2c and d). Our results show similar levels of initial DNA damage after 2 h; however, after 96 h, lower numbers of cells exhibiting DNA breaks were observed in PKCη-knockdown cells, suggesting that the response to DNA damage (repair processes) was more efficient in these cells.
PKCη exhibits opposing effects on IL-6 and IL-8 transcription and secretion: it enhances IL-6 but suppresses IL-8
Senescence is accompanied by enhanced secretion of cytokines, growth factors and proteases-collectively defining the SASP. Components of the senescence secretome reinforce or implement cell-cycle arrest and contribute to tumor suppression by signaling and recruiting the immune system to eliminate senescent cells.10 Among these, IL-6 secretion was implicated in reinforcing senescence5, 40 but also in promoting tumor invasiveness.41
We have initially determined the transcription of cytokines in PKCη-knockdown cells using RT-PCR in response to oxidative stress and chemotherapy (Figure 3). mRNA levels of all cytokines examined, that is, IL-6, IL-8, tumor necrosis factor alpha (TNF-α), vascular endothelial growth factor (VEGF) and insulin-like growth factor-binding protein (IGFBP-7), were increased, demonstrating their induction in senescence (data not shown). However, PKCη-knockdown had an effect on IL-6 and IL-8 (Figure 3) but not on the transcription of other cytokines. Oxidative stress markedly increased IL-6 mRNA expression in scrambled control cells (sevenfold) compared with its increase in PKCη-knockdown cells (twofold). Etoposide had a similar effect on IL-6 expression (Figure 3a). Notably, PKCη-knockdown cells exhibited higher levels of IL-8 secretion in response to oxidative stress and chemotherapy (30–40-fold increase) compared with PKCη-expressing scrambled control cells (10-fold increase). Thus, our studies suggest that PKCη is a negative regulator of IL-8 transcription (Figure 3b).

PKCη expression has opposing effects on IL-6 and IL-8 transcription in response to oxidative stress and DNA damage. MCF-7 clones expressing sh-PKCη (sh2-2 and sh3-3) or scrambled (sh5-3) control cells were treated with 150 μM H2O2 or 400 μM etoposide for 2 h, followed by the addition of fresh medium for 96 h. Total RNA was extracted and cDNA was generated by reverse transcription as described in Materials and Methods. RNA products were analyzed by real-time PCR according to the manufacturer’s instructions. (a) IL-6 mRNA and (b) IL-8 mRNA levels are depicted. Columns represent mean±S.E. of three independent experiments (^ indicates statistical significance compared with untreated cells, * indicates statistical significance compared with other clones). Two-tailed, unpaired sample t-test statistical analysis is shown: */^P≤0.05, **/^^P≤0.001 and ***/^^^P≤0.0001
Next, we have examined whether the opposite effects of PKCη on the transcription of IL-6 and IL-8 are consistent with their secretion (Figure 4). IL-6 secretion was markedly increased in PKCη expressing cells (5–6-fold increase) compared with PKCη-knockdown clones (1.2–2-fold increase) (Figure 4a). A similar secretion pattern was seen when these cells were exposed to etoposide (Figure 4b). Moreover, when PKCη cDNA was re-introduced on the background of the sh2-2 knockdown clone, it was able to rescue IL-6 secretion (see Supplemental Data Figure 1S). Higher levels of IL-8 were secreted from PKCη-knockdown cells compared with PKCη-expressing scrambled control cells both under basal conditions and in response to oxidative stress and etoposide (Figures 4c and d). Taken together, our results show opposing effects of PKCη on IL-6 and IL-8 secretion; PKCη enhances IL-6, but suppresses IL-8 secretion.

PKCη expression has opposing effects on IL-6 and IL-8 secretions. MCF-7 clones expressing sh-PKCη (sh η 3-5, sh η 2-2 and sh η 4-2) or scrambled control cells (sh scr 5-3) were treated with 150 μM H2O2 or 400 μM etoposide for 2 h. Fresh growth medium was added for 96 h. Medium was collected and assayed by ELISA for (a, b) IL-6 and (c, d) IL-8 protein levels as described in Materials and Methods. Values are represented as pg/ml, calculated from standard curves of at least three separate experiments performed in triplicate. Error bars represent the S.D.; two-tailed, unpaired sample t-test statistical analysis is shown: *P≤0.05, **P≤0.001 and ***/$$$P≤0.0001 (* represents statistical significance for each non-treated and treated clone and $ represents statistical significance between different clones)
PKCη is involved in an amplification loop that enhances IL-6 production
To complement the experiments shown above demonstrating a role for PKCη in IL-6 transcription (Figure 3), we overexpressed PKCη cDNA in MCF-7 cells along with the empty control vector (pHACE) (Figure 5). Doxorubicin treatment increased IL-6 transcription in control vector transfected MCF-7 cells by 12-fold, while there was a 50-fold increase in IL-6 transcription with overexpression of PKCη (Figure 5a). IL-6 receptor (IL-6R) mRNA levels were also elevated with overexpression of PKCη. (Figure 5b). IL-6 was previously shown to reinforce senescence.42 Thus, our studies suggest that PKCη acts on both IL-6 and its receptor to create an amplification loop for IL-6 production that could act in an autocrine manner to intensify senescence.

Overexpression of PKCη in MCF-7 cells increases transcription of IL-6 and IL-6R upon DNA damage. MCF-7 cells were transfected with PKCϖ cDNA and the control vector pHACE. The cells were treated with 2 μM doxorubicin for 2 h, followed by fresh medium for 48–96 h. Total RNA was extracted and cDNA was generated by reverse transcription as described in Materials and Methods. RNA products were analyzed by real-time PCR according to the manufacturer’s instructions. (a) IL-6 mRNA and (b) IL-6R mRNA levels are depicted. Columns represent mean±S.E. of three independent experiments (^ indicates statistical significance compared with untreated cells, *indicates statistical significance compared with other clones). Two-tailed, unpaired sample t-test statistical analysis is shown: *P≤0.05, **/^^P≤0.001 and ***/^^^P≤0.0001
The polymorphic variant V374I enhances the kinase activity of PKCη, IL-6 production, p21Cip1 expression and senescence
The PKCη polymorphic variant, 374I, in which valine replaces isoleucine at position 374 in its kinase domain (Figure 6a), was previously implicated in an increased risk for brain infarction.33 A plasmid containing this polymorphic variant was introduced into Cos-7 and MCF-7 cells (Figure 6). Results showed that this polymorphic variant exhibited significantly enhanced kinase activity, both in autophosphorylation assays and when myelin-basic protein (MBP) or myristoylated alanine-rich C-kinase substrate (MARCKS) were used as exogenous substrates (Figures 1b and c).

The PKCϖ polymorphic variant, 374I, shows higher kinase activity, increased p21Cip1, IL-6 secretion and β-gal staining compared with PKCη-WT. (a) Representation of PKCη’s structural domains and priming phosphorylation sites. The 374I polymorphism is indicated at the ATP binding site. The 374I polymorphic variant shows higher kinase activity as compared with WT-374V in (b) autophosphorylation assays and (c) exogenous substrate phosphorylation using MARCKS and MBP as substrates. Plasmid constructs (WT-374V, 374I and pHACE control vector) were transiently expressed in COS-7 cells. Immunoprecipitation and kinase assays using PMA for activation were performed as described in Material and Methods. The heavy IgG chain shows equal loading by Ponceau staining. The results represent five different experiments. (d) PKCη and the polymorphic variant enhance senescence markers. MCF-7 cells were transfected with WT-374V, 374I and the pHACE control vector and treated with H2O2 to induce senescence, as described above. The cells were lysed and the senescence markers p21Cip1, p16INK4a and pRB expression were detected using specific antibodies. β-Actin was used as a marker for equal protein loading. Results shown are representative of three independent experiments. (e) IL-6 production is dependent on the kinase activity of PKCη. MCF-7 cells were transfected and treated with H2O2 to induce senescence as described above. IL-6 secretion levels were measured using ELISA. (f) Overexpression of both PKCη and the polymorphic variant enhances SA-β-gal staining induced by oxidative stress. MCF-7 cells were transfected and treated as above. Cells were stained for both Hoechst and SA-β-gal 96 h after oxidative stress treatment. The total cell number was determined by nuclear fluorescent staining. Error bars represent the S.D.; two-tailed, unpaired sample t-test statistical analysis is shown: *P≤0.05, **/$$P≤0.001 and ***P≤0.0001 (* represents statistical significance for each non-treated and treated clone and $ represents statistical significance between different clones)
We then examined whether this polymorphic change would affect the secretion of IL-6. Indeed, the variant V374I exhibited increased IL-6 secretion when expressed in MCF-7 cells compared with WT-374V (Figure 6e). This could not be the result of higher 374I expression, since 374I and 374V were expressed equally (Figure 6d).
Our present studies, using sh-RNA PKCη-knockdown cells, showed that PKCη had a role in the enhanced expression of the cell cycle inhibitor, p21Cip1 (Figure 2). Here we show that overexpression of both 374V and 374I in MCF-7 cells increased the expression of the senescence marker p21Cip1 under oxidative stress conditions, with the polymorphic variant 374I having a greater effect (Figure 6d). The mutation of the INK4a/ARF locus in MCF-7 cells43 may explain the weak effect exhibited on p16INK4a expression in these cells. Phosphorylation of Rb protein was decreased under oxidative stress conditions, consistent with the fact that these cells stopped proliferating. Furthermore, the numbers of β-gal-positive cells detected in cells overexpressing the 374I variant were higher compared with that of WT or empty vector transfected cells (Figure 6f). Taken together, our studies suggest that the kinase activity of PKCη is important in promoting senescence and its maintenance.
Discussion
Cellular senescence appears to be an important mechanism preventing the proliferation of potential cancer cells. The senescence program impacts the senescence-associated cell-cycle arrest and the tissue microenvironment.11 Here we show that PKCη promotes senescence induced by oxidative stress and chemotherapy via two mechanisms; one is the upregulation of p21Cip1 and p27Kip1 that are crucial for cell cycle inhibition. The other is the increased secretion of both IL-6 and its receptor, forming a positive loop for enhanced IL-6 secretion, which could establish and further reinforce the senescence phenotype. The role of PKCη in enhancing IL-6 secretion while inhibiting IL-8 secretion could have important implications on the recruitment of immune cells to the senescent microenvironment. Hence, PKCη could modulate secretome properties. Moreover, the role of PKCη in promoting senescence is further demonstrated by employing its 374I polymorphic variant (in the ATP binding domain). This polymorphic variant, exhibiting higher kinase activity compared with WT-374V, was also more effective in promoting the secretion of IL-6, increasing p21Cip1 and augmenting senescence.
Senescent cells express a SA-β-gal which partly reflects the increase in lysosomal mass.44 Our results show that the percentage of senescent cells identified by β-gal staining in response to oxidative stress or chemotherapy-induced senescence was significantly diminished when PKCη was knocked down. Moreover, re-plated senescent cultures of PKCη-knockdown cells exhibited higher proliferation and colony formation capacity compared with PKCη-expressing control cells (Figures 1b–d), in accordance with a smaller fraction of senescent cells present in the PKCη-knockdown cells.
Cellular senescence pathways are believed to have multiple layers of regulation. Among the cellular pathways reported to regulate senescence are the p16INK4a/pRB pathway, the p19ARF/p53/p21CIP1/WAF1 pathway and the PTEN/p27KIP1 pathway.45 p21Cip1 has direct inhibitory actions on the cell-cycle machinery and correlates well with declining growth rates in cultures undergoing senescence. Indeed, induction of p21Cip1 causes cell-cycle arrest in senescent cells.46 p27Kip1 was previously shown to promote senescence in multiple tissues and loss of p27Kip1 expression led to downregulation of senescence and progression of cancer.47, 48 Moreover, an intrinsic cooperation between p21Cip1 and p27Kip1 in the activation of stress-induced senescence and tumor progression was demonstrated in vivo.49
According to our previous studies, PKCη has a role in cell cycle regulation. The expression of the cyclin-dependent kinase inhibitor p21Cip1 was specifically elevated in PKCη-expressing cells.20 In addition, p21Cip1 associated with the cyclin E/Cdk2 complex in cells under stress of serum starvation.22 Moreover, p21Cip1 was shown to associate with the PKCη/Cyclin E/Cdk2 complex in keratinocytes and to be phosphorylated by PKCη.21 The functional consequence of this phosphorylation has not been elucidated.
Our results presented here suggest that PKCη plays a role in the upregulation of p21Cip1 and p27Kip1 in senescence, as we show a marked decrease in p21Cip1 and p27Kip1 expression in PKCη-knockdown cells (Figure 2). Although PKCη increased p21Cip1 expression, it had no effect on p53 levels in treated cells (data not shown), suggesting that the upregulation of p21Cip1 is p53 independent. Further studies should be conducted to determine whether p21Cip1 is directly phosphorylated by PKCη under conditions that lead to senescence. Decreased p-AKT activity was shown to promote cellular senescence through upregulation of p53 and p27Kip1 expression.50 We have previously shown that activated PKCη reduces phosphorylation of AKT induced by insulin-like growth factor 1,26 which could be responsible for the increase in p27Kip1. Alternatively, the increase in p21Cip1 and p27Kip1 could result from the effects of PKCη on the transcription and secretion of IL-6 shown here (Figures 3,4,5,6), since IL-6 was previously shown to upregulate p27Kip1 and p21Cip1, leading to cell-cycle arrest.51, 52
Various stimuli inducing senescence often activate DDR pathways involved in both the induction and maintenance of senescence.53 Our results show reduced γ-H2AX phosphorylation, one of the DDR markers, in senescent cultures of PKCη-expressing cells (Figure 2). We have previously reported that PKCη protects against cell death induced by DNA damage via inhibition of c-Jun N-terminal kinases (JNK) activity.26 H2AX was found to be a target of the JNK signaling pathway,54 which could explain its reduced phosphorylation in PKCη-expressing control cells. Direct analysis of double strand DNA breaks by the comet assay showed more DNA breaks in PKCη-expressing control cells subsequent to initial equal DNA damage (Figures 2c and d), suggesting a reduced cellular response to DNA damage in PKCη-expressing control cells. This point needs further investigation.
Senescent cells secrete a complex mixture of extracellular proteins and soluble factors, known as the SASP.55 nuclear factor kappa B (NFκB) was shown to be a master regulator of SASP, by influencing the expression of NFκB target genes. Proteomic analysis of senescent chromatin identified the NFκB subunit p65 as a major transcription factor that accumulates on chromatin of senescent cells.56 We have recently reported that PKCη regulates NFκB upstream signaling by activating the IkB kinase and the degradation of IkB. Furthermore, PKCη enhanced nuclear translocation and transactivation of NFκB under non-stressed conditions and in response to DNA damage by camptothecin.24 Thus, PKCη could exert its effects on SASP through its role in NFκB activation in these cells.24
Components of the senescence secretome reinforce cell-cycle arrest and contribute to tumor suppression by signaling and recruiting components of the immune system10. Here we demonstrate that PKCη expression modulates the senescence secretome, mainly by inducing the transcription and secretion of IL-6 and suppressing IL-8 (Figures 3 and 4). Although the mRNA levels of other SASP components, such as TNFα, IGFBP-7 and VEGF, were induced upon oxidative stress or chemotherapy, their expression was not affected by PKCη (data not shown). IL-6 was found to be a key factor in generating cell-cycle arrest characteristics of senescence.5, 57 Moreover, IL-6 and its receptor, IL-6Ra, form a senescence-inducing circuit.42 For example, IL-6 was reported to act in concert with its receptor to induce cell-cycle arrest in response to BRAF oncogenic stress.11 Our studies show that the overexpression of PKCη in MCF-7 cells induced the expression of both IL-6 and IL-6R (Figure 5), suggesting that this circuit could be the underlying mechanism for the enhanced IL-6 expression in PKCη expressing cells in senescence.
PKCη expression resulted in the suppression of IL-8 mRNA transcription and secretion, which was alleviated when PKCη was knocked down (Figures 3 and 4). Secretion of IL-8 was frequently detected in senescent cells; however, in contrast to IL-6, its specific contribution to senescence has not been established.58 The expression of IL-8 in breast cancer cell lines was shown to require a complex cooperation between NFκB, AP-1 and C/EBP transcription factors59. Binding sites for these transcription factors were identified in the IL-6 promoter region60. Although NFκB is crucial for IL-8 gene transcription, its cooperation with either AP-1 or C/EBP is required to fully activate IL-8 transcription in breast cancer cells.59 Moreover, it was demonstrated that IL-6 expression is regulated by NFκB, while the regulation of IL-8 more closely correlated with AP-1 activity.61 Several studies demonstrated that inhibition of PKC blocks NFκB and AP-1 activity, suggesting direct regulation of these transcription factors by PKC.62 Furthermore, IL-8 release was downregulated by inhibition of JNK activity.61 Our results demonstrating that PKCη is a repressor of IL-8 are in accordance with the notion that AP-1 is important for its regulation, since we have previously shown that PKCη inhibits JNK activity.26 Thus, in the absence of PKCη, JNK will enhance the expression of IL-8 through AP-1 binding and activation of the IL-8 promoter. IL-8 is a proinflammatory molecule that functions within the tumor microenvironment. It acts on leukocytes and endothelial cells, via their CXCR1 and CXCR2 receptors to promote immune infiltration and angiogenesis.58 Furthermore, signaling through CXCR2 leads to senescence in p53-WT and non-transformed cells.57
In summary, our present study demonstrates that PKCη has a role in establishing the senescence phenotype and the presence or absence of PKCη has a profound effect on SASP. PKCη expression suppresses the expression of IL-8 but activates a positive loop for IL-6 expression. Hence, PKCη reinforces senescence through an IL-6 dependent autocrine mechanism, while the suppression of IL-8 could affect the microenvironment in a paracrine manner. As there is now considerable interest in developing novel therapies to activate senescence to control tumor promotion, or therapies to eliminate senescence, it is crucial to first elucidate the molecular regulators of senescence. This will improve our ability to develop new strategies to harness senescence as a potential cancer therapy in the future.
Materials and Methods
Cell culture, antibodies, reagents and transfections
Cells were grown in Dulbecco’s Modified Eagle Medium containing 100 U/ml penicillin, 0.1 mg/ml streptomycin and 2 mM L-glutamine and 10% Fetal Bovine Serum in a 5% CO2 humidified atmosphere at 37 °C. PKCη-shRNA-expressing, stable MCF-7 derived cell lines were generated using the SureSilencing (SA Biosciences, Frederick, MD, USA) pre-designed PKCη short hairpin RNA plasmids (sh PKCη 4-2, sh PKCη 2-2, sh PKCη 3-3 and sh PKCη 3–5) or a scrambled control vector (sh scramble 5–3).
Antibodies used included: anti-PKCη (sc-215), anti p21Cip1 (sc-397), anti p16 (sc-468), anti-DcR2 (sc-7550), which were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-actin (691001, ICN Biomedicals, Irvine, CA, USA), anti-HA.11 Clone 16B12 Monoclonal (MMS-101P, Convance, Emeryville, CA, USA), anti pRB (554136, BD pharmingen, San Jose, CA, USA), anti-γ-H2AX phosphor-S139 (ab18311, Abcam, Cambridge, MA, USA) and anti-p53 (Ab-6, Calbiochem, San Diego, CA, USA). For the detection of primary antibodies, blots were incubated with horseradish peroxidase conjugated to donkey anti-rabbit/anti-mouse/ anti-goat immunoglobulin (Amersham Pharmacia, Buckinghamshire, UK). The antibodies were detected by an ECL reagent (RPN 2209, Amersham Pharmacia, New York, NY, USA).
Transfections of COS-7 and MCF-7 cells were carried out in 60 or 100 mm culture dishes 24 h post seeding. The cells were transfected with the indicated plasmids using the jetPEI reagent (PolyPlast transfection, Cedex, France) or the TurboFect transfection reagent (Fermentas, Hanover, MD, USA) according to the manufacturer’s instructions. Following transfection, cells were treated with 150 μM H2O2, 400 μM etoposide (Ebewe Pharma, Unterach, Austria) or 2 μM doxorubicin for 2 h, followed by recuperation with fresh medium for 48–96 h.
Plasmids
The GFP–PKCη-374I construct was generated using GFP–PKCη-WT that was previously constructed63. The two first PCR fragments were amplified using flanking primers A1 to mut-3′ and B1 to mut-5′ from the hPKCη cDNA. This was followed by PCR using the N- and C-terminal primers: primer A1 and primer B1, together with two overlapping fragments. The insert was digested by XbaI and HindIII and ligated into the GFP–C2 plasmid vector. Next, the completed insert was amplified by PCR using HA EcoRI insertion primers and digested with EcoRI before it was ligated into the pCDNA3/HACE cloning vector. The following primers were used for the PCR reactions: Primer mut-3′ (V374I 3′): 5′-TCCTGTTTCTTTTATTCTTGCAAGCA-3′; Primer mut-5′ (V374I 5′): 5′-TGCTTGCAAGAATAAAAGAAACAGGA-3′; Primer B1-(3′ primer): 5′-CCTTTTCTAGATCGTGCCCTCTCTCTCACT-3′; Primer A1-(5′ primer): 5′-CGAGCTCAAGCTTCGAATTCTGC-3′; EcoRI HA 3′: 5′-CATA-GAATTC- TGGTTGCAATTCTGGAGAC-3′; EcoRI HA 5′: 5′-CC- GAATTC –TCGTCTGGCACCATGAA-3′.
Immunoblot analysis
Cell lysates were prepared using RIPA lysis buffer containing 10 mM Tris-HCl pH 8.0, 100 mM NaCl, 5 mM EGTA, 0.1% SDS, 1% NP-40, 45 mM β-mercaptoethanol and 50 mM NaF. Protease inhibitors (1 mM PMSF, 100 μg/ml aprotinine and 10 μg/ml leupeptin) and phosphatase inhibitors (100 μM sodium orthovanadate, 50 mM β-glycerolphosphate and 5 mM sodium pyrophosphate) were added just before cell lysis. Lysates were placed on ice for 30 min and sheared several times through a 21-guage needle. Lysates were centrifuged at 14 000 × g for 15 min at 4 °C and protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA).
Immunoprecipitations and in vitro kinase assays
Immunoprecipitations were performed using pre-absorbed mAbs on protein A/G-agarose beads for 1 h at 4 °C. Excess Ab were removed by five washes in cold lysis buffer (see above), and Ab-coated beads were incubated with cell lysates overnight at 4 °C. Immune complexes were precipitated by centrifugation, followed by extensive washing in lysis buffer. Samples of cell lysates or immunoprecipitates were mixed with equal volumes of 2 × SDS sample buffer, incubated at 95 °C for 5 min and fractionated by SDS-PAGE. IP beads were rinsed in 1% Triton one time and then equal aliquots were transferred to Eppendorf tubes and rinsed two more times. Kinase assay buffer mix was prepared as follows: MgCl2 5 mM, CaCl2 300 μM, Tris-HCl pH 7.4 20 mM, PS 25 μmol/25 μl, PMA 1 μM or 1.6 μM was added separately to the complete buffer as needed. As a substrate, either 7 μg MBP (M1891, Sigma, St. Louis, MO, USA) or GST-MARCKS was added to the beads before the reaction mix was used. [γ-32P]ATP 4Ci/mmol was added to the mix seconds before initiating the reaction. All solutions were prepared on ice. Samples containing 20 μl beads were mixed with 23 μl kinase assay mix and incubated for either 10 or 20 min at 30 °C. The reaction was stopped by adding 10 μl sample buffer and 5 min denaturation at 95 °C, followed by spin down. All samples were run on 10% SDS-PAGE and transferred to a PVDF membrane (Bio-Rad, Herts, UK). Kinase activity was evaluated using a phosphor-imager (Personal Molecular Imager (PMI) System, Bio-Rad) and developing film exposures.
β-galactosidase staining
SA-β-gal activity was determined using a previously described protocol64 with some modifications. Briefly, cells were washed once with PBS, fixed with 0.5% glutaraldehyde (PBS (pH 7.2)), and washed in PBS (pH 7.2) supplemented with 1 mM MgCl2. Cells were stained in X-gal solution (1 mg/ml X-gal (Boehringer, Ingelheim, Germany), 0.12 mM K3Fe [CN]6, 0.12 mM K4Fe[CN]6, 1 mM MgCl2 in PBS at pH 6.0) overnight at 37 °C. Cells were photographed using an IX70 Olympus optical light microscope (Olympus, Tokyo, Japan). To estimate total cell numbers, cell cultures were stained with Hoechst 10 mg/ml (Calbiochem, HO 33342) for 30 min at 37 °C prior to β-gal staining. Hoechst florescence was detected using a light source providing light at 340–380 nm, emission was at 465 nm. SA-β-gal-positive cells were calculated as the percentage of Hoechst-stained cells.
Cytokine analysis
MCF-7 cells, transfected with PKCη-expressing plasmids (pHACE), or PKCη-shRNA stable cell lines of PKCη-knockdown cells or scrambled control cells were seeded in six-well plates (4 × 105 cells/well). After 24–48 h, the cells were treated with 150 μM H2O2, 400 μM etoposide or 2 μM doxorubicin for 2 h (as described above). Supernatants were collected from the culture medium. Cytokine levels were evaluated using the manufacturer’s standard curves. The ELISA kits used were: IL-6 and IL-8 (Cat. # 88-7066-88 and 88-8086, eBioscience, San Diego, CA, USA).
mRNA isolation, cDNA synthesis and quantitative real-time PCR
Total RNA was isolated and purified using an RNA extraction kit (5Prime, # 2900319, PerfectPure, Gaithersburg, MD, USA) according to the manufacturer’s protocol. The carried-over DNA was eliminated by treatment with DNase (Turbo DNA-free, Ambion, Foster City, CA, USA). The first-strand cDNA was reverse transcribed from 0.5–1 μg total RNA using the Reverse-iT first-strand synthesis kit and random hexamer primers (ABgene, Surrey, UK). Quantitative real-time PCR was performed using an ABI 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA). Specific mRNA levels were quantified using SYBR green reagents (Kapa Biosystems, Cape Town, South Africa): primers for IL-8 (5′-CAAGAGCCAGGAAGAAACCA-3′ (forward) and 5′-AGCACTCCTTGGCAAAACTG-3′ (reverse)); TNFα (5′ ACTTTGGAGTGATCGGCC-3′ (forward) and 5′-GTTGTAGCAAACCCTCAAGC-3′ (reverse)); IL-6 (5′-GGGCTGAACGGTCAAAGACA-3′ (forward) and 5′-GGATGACACAGTGATGCTGGA-3′ (reverse)); and VEGF (5′-CTACCTCCACCATGCCAAGTGGTCC-3′ (forward) and 5′-ATGTCCACCAGGGTCTCGATTGGA-3′ (reverse)). All genes were normalized to β-Actin using specific primers (5′-AGCCTCGCCTTTGCCGATCC-3′ (forward) and 5′-TTGCACATGCCGGAGCCGTT-3′ (reverse)) or GAPDH. The PCR thermo cycling parameters were 95 °C for 3 min and 40 cycles of 95 °C for 3 s and 60 °C for 30 s. All gene-specific primers used were examined for efficiency, displaying an amplification slope of −3.33±0.3 (r>0.98) Data analysis was performed using the cycle threshold ΔΔCT method. Statistical analysis of differences between the groups was performed as described below.
Colony formation assays
Cell cultures treated for senescence induction were harvested with trypsin-EDTA and counted using a hemocytometer. A glass Pasteur pipette was used to ensure cell separation. Cells were diluted in complete medium, and 1 × 105 cells were plated in 100 mm tissue culture dishes. After incubation for 10±14 days at 37 °C in 5% CO2, the cells were washed with PBS and stained with 0.5% Crystal Violet in glutaraldehyde for 30 min at room temperature. Culture dishes were submerged and washed in tap water three to five times. Plates were left to dry overnight. The size and density of visible colonies were evaluated.
Comet assay
PKCη-shRNA stable cell lines of PKCη-knockdown cells or scrambled control cells were seeded in six-well plates (4 × 105 cells/well). After 24–48 h, the cells were treated with 400 μM etoposide for 2 h, followed by replacement with fresh medium for 96 h. Single-cell gel electrophoresis was performed as described65. Briefly, fully frosted microscope slides were covered with 150 μl of 1.5% normal, melting-point agarose in PBS and then flattened using a cover slip for 5–10 min at 4 °C. The dried slides were kept at room temperature. Approximately 5 × 104 cells per sample (10 μl) were mixed with 0.5% (w/v) low melting agarose (120 μl) to form a cell suspension, which was then pipetted onto the first agarose layer, flattened and solidified (see above). After removal of the cover slip, the slides were immersed in the neutral lysis solution at 4 °C for 1 h or longer in the dark. The basic lysis solution prepared in advance included: 2.5 M NaCl, 100 mM EDTA, 10 mM Tris pH 10 and 1% sodium lauryl sarcosinate, and were kept at room temperature for up to 1 month. Immediately before use, 1% Triton X-100 and 10% dimethylsulphoxide were added to the basic lysis solution, mixed for ~20 min and refrigerated. The slides were incubated in electrophoresis buffer at 4 °C for 20 min and electrophoresis was performed at 25 V, 100 mA for 20 min. Next, the slides were immersed in neutralization buffer (three times for 5 min) and washed three times with DDW. The dried slides were stained with 4′,6-diamidino-2-phenylindole solution (1 μg/ml) and covered with a cover slip. A minimum of 50 different fields were photographed in three separate experiments with an IX70 Olympus optical light microscope (Olympus). The volume of comet tails of at least 250 cells per time point was assessed.
Statistical analysis
The statistical significance of differences between experimental groups was determined using the ANOVA test of variance in cases of multiple variables as well as the unpaired two-tailed sample t-test (using Prisma 5 program, Prism - Graphpad, La Jolla, CA, USA). P-value<0.05 was considered statistically significant.
Abbreviations
- CDK:
-
cyclin-dependent kinase
- DDR:
-
DNA damage response
- H2AX:
-
H2A histone family, member X
- IGFBP:
-
insulin-like growth factor-binding protein
- IL-6:
-
interleukin-6
- IL-6R:
-
interleukin-6 receptor
- JNKs:
-
c-Jun N-terminal kinases
- MARCKS:
-
myristoylated alanine-rich C-kinase substrate
- MBP:
-
myelin-basic protein
- NFκB:
-
nuclear factor kappa B
- PKC:
-
protein kinase C
- Rb:
-
retinoblastoma
- SA-β-gal:
-
senescence-associated β-galactosidase
- SASP:
-
senescence-associated secretory phenotype
- TNFα:
-
tumor necrosis factor alpha
- VEGF:
-
vascular endothelial growth factor
References
Rodier F, Campisi J . Four faces of cellular senescence. J Cell Biol 2011; 192: 547–556.
Suzuki M, Boothman DA . Stress-induced premature senescence (SIPS)—influence of SIPS on radiotherapy. J Radiat Res 2008; 49: 105–112.
Shay JW, Roninson IB . Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 2004; 23: 2919–2933.
Ewald JA, Desotelle JA, Wilding G, Jarrard DF . Therapy-induced senescence in cancer. J Natl Cancer Inst 2010; 102: 1536–1546.
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008; 133: 1019–1031.
Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM et al. BRAFE600—associated senescence-like cell cycle arrest of human naevi. Nature 2005; 436: 720.
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995; 92: 9363–9367.
Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003; 22: 4212–4222.
Kuilman T, Peeper DS . Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer 2009; 9: 81–94.
Freund A, Orjalo AV, Desprez PY, Campisi J . Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 2010; 16: 238–246.
Kuilman T, Michaloglou C, Mooi WJ, Peeper DS . The essence of senescence. Genes Dev 2010; 24: 2463–2479.
Paget JA, Restall IJ, Daneshmand M, Mersereau JA, Simard MA, Parolin DA et al. Repression of cancer cell senescence by PKCiota. Oncogene 2012; 31: 3584–3596.
Katakura Y, Udono M, Katsuki K, Nishide H, Tabira Y, Ikei T et al. Protein kinase C δ plays a key role in cellular senescence programs of human normal diploid cells. J Biochem 2009; 146: 87–93.
Oliva JL, Caino MC, Senderowicz AM, Kazanietz MG . S-Phase-specific activation of PKC alpha induces senescence in non-small cell lung cancer cells. J Biol Chem 2008; 283: 5466–5476.
Bacher N, Zisman Y, Berent E, Livneh E . Isolation and characterization of PKC-L, a new member of the protein kinase C-related gene family specifically expressed in lung, skin, and heart. Mol Cell Biol 1991; 11: 126–133.
Livneh E, Shimon T, Bechor E, Doki Y, Schieren I, Weinstein IB . Linking protein kinase C to the cell cycle: ectopic expression of PKCη in NIH-3T3 cells alters the expression of cyclins and Cdk inhibitors and induces adipogenesis. Oncogene 1996; 12: 1545–1555.
Hussaini IM, Karns LR, Vinton G, Carpenter JE, Redpath GT, Sando JJ et al. Phorbol 12-myristate 13-acetate induces protein kinase Cη-specific proliferative response in astrocytic tumor cells. J Biol Chem 2000; 275: 22348–22354.
Fishman D, Segal S, Livneh E . The role of protein kinase C in G1 and G2/M phases of the cell cycle. Int J Oncol 1998; 12: 181–186.
Livneh E, Fishman DD . Linking protein kinase C to cell cycle control. Eur J Biochem 1997; 248: 1–9.
Fima E, Shtutman M, Libros P, Missel A, Shahaf G, Kahana G et al. PKCη enhanced cell cycle progression, the expression of G1 cyclins and p21 in MCF-7 cells. Oncogene 2001; 20: 6794–6804.
Kashiwagi M, Ohba M, Watanabe H, Ishino K, Kasahara K, Sanai Y et al. PKCη associates with cyclin E/Cdk2/p21 complex, phosphorylates p21 and inhibits Cdk2 kinase in keratinocytes. Oncogene 2000; 19: 6334–6341.
Shtutman M, Hershko T, Maissel A, Fima E, Livneh E . PKCη associates with cyclin E/Cdk2 complex in serum-starved MCF-7 and NIH-3T3 cells. Exp Cell Res 2003; 286: 22–29.
Akkaraju GR, Basu A . Overexpression of protein kinase C-h attenuates caspase activation and tumor necrosis factor a -induced cell death. Biochem Biophys Res Commun 2000; 279: 103–107.
Raveh-Amit H, Hai N, Rotem-Dai N, Shahaf G, Gopas J, Livneh E . Protein kinase Ceta activates NF-kappaB in response to camptothecin-induced DNA damage. Biochem Biophys Res Commun 2011; 412: 313–317.
Tamarkin A, Zurgil U, Braiman A, Hai N, Krasnitsky E, Maissel A et al. DNA damage targets PKCh to the nuclear membrane via its C1B domain. Exp Cell Res 2011; 10: 1465–1475.
Rotem-Dai N, Oberkovitz G, Abu-Ghanem S, Livneh E . PKCeta confers protection against apoptosis by inhibiting the pro-apoptotic JNK activity in MCF-7 cells. Exp Cell Res 2009; 315: 2616–2623.
Abu-Ghanem S, Oberkovitz G, Benharroch D, Gopas J, Livneh E . PKCη expression contributes to the resistance of Hodgkin's lymphoma cell lines to apoptosis. Cancer Biol Ther 2007; 6: 1375–1380.
Beck J, Bohnet B, Brugger D, Bader P, Dietl J, Scheper RJ et al. Mutiple gene expression analysis reveals distinct differences between G2 and G3 stage breast cancers, and correlations of PKCη with MDR1, MRP and LRP gene expression. Br J Cancer 1998; 77: 87–91.
Beck JF, Bohnet B, Brugger B, Dietel J, Scheper RJ . Expression analysis of protein kinase C isoenzymes and multidrug resistance associated genes in ovarian cancer cells. Anticancer Res 1998; 18: 701–705.
Beck JF, Handgretinger R, Klingebiel T, Dopfer R, Schaich M, Ehninger G . Expression of PKC isozyme and MDR-associated genes in primary and relapsed state AML. Leukemia 1996; 10: 426–433.
Karp G, Abu-Ghanem S, Novak V, Mermerstain W, Ariad S, Sion-Vardy N et al. Localization of PKCη in cell membranes as a predictor for breast cancer response to treatment. Onkologie 2012; 35: 260–266.
Krasnitsky E, Baumfeld Y, Freedman J, Sion-Vardy N, Ariad S, Novack V et al. PKCη is a novel prognostic marker in non-small cell lung cancer. Anticancer Res 2012; 32: 1507–1514.
Kubo M, Hata J, Ninomiya T, Matsuda K, Yonemoto K, Nakano T et al. A nonsynonymous SNP in PRKCH (protein kinase C eta) increases the risk of cerebral infarction. Nat Genet 2007; 39: 212–217.
Takata Y, Hamada D, Miyatake K, Nakano S, Shinomiya F, Scafe CR et al. Genetic association between the PRKCH gene encoding protein kinase Ceta isozyme and rheumatoid arthritis in the Japanese population. Arthritis Rheum 2007; 56: 30–42.
Wu L, Shen Y, Liu X, Ma X, Xi B, Mi J et al. The 1425G/A SNP in PRKCH is associated with ischemic stroke and cerebral hemorrhage in a chinese population. Stroke 2009; 40: 2973–2976.
Goto Y, Hishida A, Matsuo K, Tajima K, Morita E, Naito M et al. PRKCH gene polymorphism is associated with the risk of severe gastric atrophy. Gastric Cancer 2010; 13: 90–94.
Byun HO, Han NK, Lee HJ, Kim KB, Ko YG, Yoon G et al. Cathepsin D and eukaryotic translation elongation factor 1 as promising markers of cellular senescence. Cancer Res 2009; 69: 4638–4647.
Dasari A, Bartholomew JN, Volonte D, Galbiati F . Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res 2006; 66: 10805–10814.
Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C et al. Senescence of activated stellate cells limits liver fibrosis. Cell 2008; 134: 657–667.
Acosta JC, Gil J . Senescence: a new weapon for cancer therapy. Trends Cell Biol 2012; 22: 211–219.
Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008; 6: 2853–2868.
Kojima H, Inoue T, Kunimoto H, Nakajima K . IL-6-STAT3 signaling and premature senescence. JAKSTAT 2013; 2: e25763.
Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995; 55: 4525–4530.
Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006; 5: 187–195.
Ben-Porath I, Weinberg RA . The signals and pathways activating cellular senescence. Int J Biochem Cell Biol 2005; 37: 961–976.
Roninson IB . Tumor cell senescence in cancer treatment. Cancer Res 2003; 63: 2705–2715.
Alexander K, Hinds PW . Requirement for p27(KIP1) in retinoblastoma protein-mediated senescence. Mol Cell Biol 2001; 21: 3616–3631.
Majumder PK, Grisanzio C, O'Connell F, Barry M, Brito JM, Xu Q et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell 2008; 14: 146–155.
Garcia-Fernandez RA, Garcia-Palencia P, Suarez C, Sanchez MA, Gil-Gomez G, Sanchez B et al. Cooperative role between p21cip1/waf1 and p27kip1 in premature senescence in glandular proliferative lesions in mice. Histol Histopathol 2014; 29: 397–406.
Zhu B, Ferry CH, Blazanin N, Bility MT, Khozoie C, Kang BH et al. PPARbeta/delta promotes HRAS-induced senescence and tumor suppression by potentiating p-ERK and repressing p-AKT signaling. Oncogene 2013; e-pub ahead of print 11 November 2013 doi:10.1038/onc.2013.477.
Flørenes VA, Lu C, Bhattacharya N, Rak J, Sheehan C, Slingerland JM et al. Interleukin-6 dependent induction of the cyclin dependent kinase inhibitor p21WAF1/CIP1 is lost during progression of human malignant melanoma. Oncogene 1999; 18: 1023–1032.
Mori S, Murakami-Mori K, Bonavida B . Interleukin-6 induces G1 arrest through induction of p27(Kip1), a cyclin-dependent kinase inhibitor, and neuron-like morphology in LNCaP prostate tumor cells. Biochem Biophys Res Commun 1999; 257: 609–614.
Salama R, Sadaie M, Hoare M, Narita M . Cellular senescence and its effector programs. Genes Dev 2014; 28: 99–114.
Lu C, Zhu F, Cho YY, Tang F, Zykova T, Ma WY et al. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell 2006; 23: 121–132.
Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 2009; 11: 973–979.
Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev 2011; 25: 2125–2136.
Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008; 133: 1006–1018.
Gales D, Clark C, Manne U, Samuel T . The chemokine CXCL8 in carcinogenesis and drug response. ISRN Oncol 2013; 2013: 859154.
Freund A, Jolivel V, Durand S, Kersual N, Chalbos D, Chavey C et al. Mechanisms underlying differential expression of interleukin-8 in breast cancer cells. Oncogene 2004; 23: 6105–6114.
Yang Y, Pares-Matos EI, Tesmer VM, Dai C, Ashworth S, Huai J et al. Organization of the promoter region of the human NF-IL6 gene. Biochim Biophys Sin Acta 2002; 1577: 102–108.
Khalaf H, Jass J, Olsson PE . Differential cytokine regulation by NF-kappaB and AP-1 in Jurkat T-cells. BMC Immunol 2010; 11: 26.
Berry N, Nishizuka Y . Protein kinase C and T cell activation. EurJ Biochem 1990; 189: 205–214.
Maissel A, Marom M, Shtutman M, Shahaf G, Livneh E . PKCη is localizes in the Golgi, ER and nuclear envelope and translocates to the nuclear envelope upon PMA activation and serum-starvation: C1b domain and the pseudosubstrate containing fragment target PKCη to the Golgi and the nuclear envelope. Cell Signal 2006; 18: 1127–1139.
Sagiv A, Biran A, Yon M, Simon J, Lowe SW, Krizhanovsky V . Granule exocytosis mediates immune surveillance of senescent cells. Oncogene 2013; 32: 1971–1977.
Speit G, Hartmann A . The comet assay (single-cell gel test). A sensitive genotoxicity test for the detection of DNA damage and repair. Methods Mol Biol 1999; 113: 203–212.
Acknowledgements
This work was supported by the Israel Science Foundation (grant No. 1413/10).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Diederich
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Zurgil, U., Ben-Ari, A., Atias, K. et al. PKCη promotes senescence induced by oxidative stress and chemotherapy. Cell Death Dis 5, e1531 (2014). https://doi.org/10.1038/cddis.2014.481
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.481
This article is cited by
-
SIRT3/6: an amazing challenge and opportunity in the fight against fibrosis and aging
Cellular and Molecular Life Sciences (2024)
-
Epigenetic signatures of gestational diabetes mellitus on cord blood methylation
Clinical Epigenetics (2017)
-
Epithelial cell senescence: an adaptive response to pre-carcinogenic stresses?
Cellular and Molecular Life Sciences (2017)
-
Downregulation of peroxiredoxin-3 by hydrophobic bile acid induces mitochondrial dysfunction and cellular senescence in human trophoblasts
Scientific Reports (2016)
-
Premature aging/senescence in cancer cells facing therapy: good or bad?
Biogerontology (2016)
