
Abstract
Mycobacterium tuberculosis infection generates pulmonary granulomas that consist of a caseous, necrotic core surrounded by an ordered arrangement of macrophages, neutrophils and T cells. This inflammatory pathology is essential for disease transmission and M. tuberculosis has evolved to stimulate inflammatory granuloma development while simultaneously avoiding destruction by the attracted phagocytes. The most abundant phagocyte in active necrotic granulomas is the neutrophil. Here we show that the ESAT-6 protein secreted by the ESX-1 type VII secretion system causes necrosis of the neutrophils. ESAT-6 induced an intracellular Ca2+ overload followed by necrosis of phosphatidylserine externalised neutrophils. This necrosis was dependent upon the Ca2+ activated protease calpain, as pharmacologic inhibition prevented this secondary necrosis. We also observed that the ESAT-6 induced increase in intracellular Ca2+, stimulated the production of neutrophil extracellular traps characterised by extruded DNA and myeloperoxidase. Thus we conclude that ESAT-6 has a leukocidin function, which may facilitate bacterial avoidance of the antimicrobial action of the neutrophil while contributing to the maintenance of inflammation and necrotic pathology necessary for granuloma formation and TB transmission.
Similar content being viewed by others

Main
Tuberculosis (TB) caused by Mycobacterium tuberculosis remains a leading source of mortality by infectious disease, with one-third of the world’s population infected, 8.6 million new cases of TB and 1.3 million deaths annually.1 The fundamental feature of TB transmission is the generation of a pulmonary tubercle lesion that contains a cuff of immune cells surrounding a necrotic core laden with extracellular bacteria. This lesion may ‘cavitate’ into the airways of the lung releasing the bacteria to allow transmission via the respiratory route. The essential contribution of macrophage and neutrophil cell death to the generation of this pathology has been recognised in many studies; however, notably in a comprehensive study of TB lesion development, Medlar2 observed that the polymorphonuclear cells were attracted to lesions following the death of mononuclear cells and that the bulk of necrotic tissue in human caseating tubercle lesions represented dead polymorphonuclear cells.
We now know that the bacterium has evolved mechanisms to regulate the mode and timing of macrophage cell death.3, 4, 5, 6, 7 After initial infection into the lungs, it is supposed that the bacterium is phagocytosed by alveolar macrophages which migrate into the interstitium of the lung.8 The bacterium is able to replicate intracellularly in the macrophage, inhibiting apoptosis until at a certain bacterial load it induces necrosis of the macrophage.9 The ensuing inflammation attracts monocytes and neutrophils from post-capillary venules that engulf the released bacteria, and thus sequential rounds of replication and inflammation enable the generation of the tubercle lesion. However, although we are beginning to understand the mechanisms of macrophage cell death control,4, 5, 6 we know very little about how M. tuberculosis modulates neutrophil death.
It is also clear that in some circumstances neutrophils have an antimycobacterial capacity,10,11 which may be mediated by the direct generation of reactive oxygen species (ROS) or by apoptosis of the infected neutrophils and subsequent efferocytosis of the apoptotic body combined with ROS-dependent killing.12,13 Additionally, neutrophil apoptosis has been linked to effective generation of adaptive immunity in M. tuberculosis infection.14 However, to counter this, M. tuberculosis has been recently shown to inhibit neutrophil apoptosis14 and furthermore, has been observed to induce necrosis.11,12 Interestingly, neutrophil necrosis only occurs on exposure to virulent strains which express the region of difference 1 (RD1) which encodes a type VII secretion system (ESX) that secretes proteins including the abundant early secretory antigen-6 (ESAT-6).12,15, 16, 17 Thus the bacterial induction of pro-inflammatory neutrophil necrosis may have dual benefit to the pathogen by removing the antimicrobial threat of the neutrophil while simultaneously facilitating the generation of the necrotic cavitating lesions that drive TB transmission.
The mechanism of necrosis in neutrophils can be varied and controlled. The most recent to be described is ‘NETosis’,18,19 whereby death of the neutrophil results in formation of a structure made of DNA with a histone backbone which contains neutrophil elastase, myeloperoxidase (MPO) and metalloproteinases. These ‘traps’ are known to be produced in vivo and associate with bacteria in some infections.18,20, 21, 22 Importantly they have been shown to be produced by M. tuberculosis infected neutrophils in vitro23 although with no bactericidal activity. As well as ‘NETosis’ there are also other described mechanisms of neutrophil death, one of which is ‘secondary necrosis’. In vitro aging, without any stimuli, results in the necrosis of neutrophils that have undergone apoptosis (termed secondary necrosis). Previous investigations have shown that neutrophils that have externalised phosphatidylserine, and are therefore ‘apoptotic’, can undergo secondary necrosis caused by a Ca2+ influx leading to the activation of a subtype of Ca2+ activated protease, calpain.24 This we termed Ca2+ Induced Necrosis (CAIN). In the present study, we elucidate the molecular events that link the RD1 encoded ESX-1 type VII secretion system with neutrophil necrosis. We chose to investigate the ESAT-6 protein, which is secreted by ESX-1, because it interacts with lipid membranes and is thought to be pore-forming,25, 26, 27, 28 thus it has the potential to influence intracellular Ca2+. Furthermore, calpain has been shown previously to be active in M.bovis infections that were dependent on the RD1 locus.29 ESAT-6 has also been shown to have cytotoxic effects to pneumocytes30 and T lymphocytes.31 Therefore, this study aimed to determine if ESAT-6 had a leukocidin action, and if so, whether this was dependent on intracellular flux of Ca2+, activation of calpain, and further, if this resulted in the formation of neutrophil extracellular traps (NETs).
Results
ESAT-6 protein increases intracellular Ca2+ and subsequent necrosis in phosphatidylserine externalised neutrophils
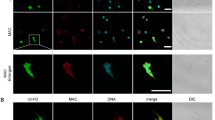
Previous work had identified increased intracellular Ca2+ as a mechanism of secondary necrosis in human neutrophils.24 To examine the effect of ESAT-6 on neutrophil Ca2+ we loaded 24- h aged cells with fluo4-AM, a fluorescent Ca2+ indicator that stays within intact cells following uptake and conversion from its AM ester to the hydrophilic fluo4 molecule.32 Using confocal microscopy we observed that the addition of ESAT-6 caused an increase in intracellular Ca2+ levels in a proportion of neutrophils within 20 min of exposure. The sequential micrographs in Figure 1a illustrate the Ca2+ influx as increasing Fluo4 fluorescence (green) in a phosphatidylserine externalised (Annexin V positive, purple) cell that ultimately undergoes necrosis (propidium iodide, red) at 60 min post-ESAT-6 addition. A movie of this response is available (Supplementary Figure 1). A representative quantitation of the Ca2+ increase followed by simultaneous Ca2+ release and uptake of propidium iodide at the point of cell lysis is shown in Figure 1b. Interestingly ESAT-6 did not induce significant calcium uptake in cells without externalised phosphatidylserine (annexin V negative) (Figure 1c).

Intracellular Ca2+ increases in ESAT-6 treated neutrophils and precedes necrosis. (a) Sequential fluorescence micrographs showing accumulation of calcium (green, Fluo4) in Annexin V positive (purple)/PS externalised aged neutrophils after exposure to exogenous ESAT-6 (20 μg/ml). Uptake of propidium iodide (PI) (red) indicates necrosis of the cell. Arrow tracks an individual cell. White bar=10 μm. Panel (b) shows a representative quantitation of intracellular Ca2+ levels of a PS externalised (Annexin V+ve) cell exposed to ESAT-6 (16 responses in N=3 experiments). Panel (c) shows representative intracellular Ca2+ in a non-PS externalised neutrophil exposed to ESAT-6. (21 responses in N=4)
To quantify the level of necrosis, we used flow cytometry to measure the percentage of propidium iodide positive cells 60 min after addition of ESAT-6 (Figure 2a). The increase in necrosis was dose dependent with 20 μg/ml ESAT-6 causing a 25% increase in necrosis and 40 μg/ml inducing 100% increase in neutrophil necrosis. As a positive control we treated neutrophils with the Streptomyces conglobatus ionophore, ionomycin, which is known to induce necrosis in neutrophils. Ionomycin induced a 150% increase in necrosis. Stimulation of aged neutrophils with a control recombinant M. tuberculosis protein (Rv0435c, chosen as a non-RD1/ESX-1 protein to control for non-specific effects of using a heterologously expressed protein) did not induce neutrophil necrosis. Further analysis by flow cytometry confirmed that necrosis was occurring preferentially in the annexin V positive neutrophils (Figure 2b). Thus we conclude that ESAT-6 causes necrosis of phosphatidylserine externalised neutrophils in a Ca2+ dependent manner.

ESAT-6 causes selective necrosis of phosphatidylserine externalised neutrophils. (a) Addition of ESAT-6 causes an increase in the proportion of propidium iodide positive cells as assessed by flow-cytometry. Control protein is recombinant Rv0435c. Ionomycin is a known inducer of neutrophil necrosis. Data normalised to vehicle in each experiment. Accumulated data N>4 for each condition (three repeats per experiment). *P<0.05 using Student’s unpaired t test compared to control protein. (b) The increase in necrosis caused by ESAT-6 (20 μg/ml) occurred preferentially in PS externalised cells (Annexin V-FITC, AnV+ve). N=4 (in triplicate). *P<0.05 using unpaired Student’s t test. Error bars are S.E.M.
Calpain activation by Ca2+ influx causes necrosis in ESAT-6 stimulated phosphatidylserine externalised neutrophils
The Ca2+ activated cysteine proteases known as calpains have been previously implicated in the necrosis of neutrophils.24 To test if calpains were involved in necrosis of ESAT-6 stimulated neutrophils, we pre-incubated aged neutrophils with the pan-specific calpain inhibitor, PD150606,33 before stimulation with ESAT-6. The inhibitor significantly reduced the level of cell death induced by ESAT-6 (Figure 3a). Furthermore, the inhibitor reduced necrosis in those cells with externalised phosphatidylserine (Figure 3b). These results indicate that ESAT-6 can induce necrosis in the phosphatidylserine externalised population through the Ca2+ activated calpain family of proteases.

Calpain mediates ESAT-6 induced neutrophil necrosis. (a) Pre-treatment of neutrophils with the calpain inhibitor, PD150606, inhibits the induction of necrosis by ESAT-6 (20 μg/ml). Necrosis was measured by flow-cytometry of propidium iodide uptake 1 h post-ESAT-6 stimulation. Data are normalised to vehicle plus DMSO. N=4 (in triplicate). *P<0.05. (b) The decrease in ESAT-6 dependent necrosis caused by calpain inhibition occurred preferentially in PS externalised cells (Annexin V-FITC, AnV +ve). Representative data N=3 (in triplicate). *P<0.05 using Student’s t test
Necrosis by ESAT-6 leads to the formation of NET-like structures
NET formation has often been seen during necrosis of neutrophils, so we tested whether NETs were produced during neutrophil necrosis induced by ESAT-6. We used confocal scanning laser microscopy to analyse the morphology and composition of the ESAT-6 treated and observed that they had an expanded structure (Figure 4) and stained positively with propidium iodide, indicative of extruded DNA consistent with previous observations of NETs (Figures 4a and b). The ESAT-6 treated cells had a larger diameter compared to cells treated with vehicle alone (Figure 4d). NETs are also known to contain MPO,34 so to further confirm the identity of the ESAT-6 induced necrotic population we assessed the presence of exposed MPO using confocal microscopy (Figure 4c) and flow cytometry (Figure 4e) with FITC labelled antibodies to MPO. This showed that MPO was widely accessible and associated with the ESAT-6-induced NET-like cell population. This localisation of MPO to the ESAT-6 NETs was similar to that observed when neutrophils were treated with ionomycin (Figure 4f). Ionomycin stimulates calcium influx across the cell membrane and thus ESAT-6 and ionomycin may stimulate NETosis through similar pathways.

ESAT-6 dependent lysis releases NETs. (a) Neutrophils were treated with 40 μg/ml ESAT-6 and the morphology and propidium iodide (PI) fluorescence (b) assessed by confocal microscopy (white bar=10 μm). Propidium iodide positive cells have an expanded morphology consistent with lysis and extrusion of NET-like structures containing DNA. (c) MPO immunoreactivity of ESAT-6 lysed neutrophils confirms identity as NETs. (d) The mean diameter of necrotic ESAT-6 stimulated neutrophils is significantly greater than necrotic cells from cultures treated with vehicle alone (P<0.001 by T test, cells counted from greater than 10 fields of view). Flow cytometric quantitation of MPO externalisation in ESAT-6 treated neutrophils (e) shows similar high levels of MPO release into the NET as that observed with ionomycin (5 μM) generated NETs (f); anti-MPO FITC (blue) compared with isotype control (green). Representative data from two donors
Discussion
The ESX-1 secretion system is an essential requirement for M. tuberculosis virulence. Its deletion from the genome of M. bovis BCG represents a key step in the attenuation of the vaccine strain and despite being perhaps the most studied molecular system in mycobacteria, its functions remain elusive but may be many.35 It secretes a number of proteins, the most prominent of which are ESAT-6 and its putative partner Cfp10, with which it forms a heterodimer.36 If there is one predominant theme in the proposed functions of ESAT-6 it is that it interacts with lipid membranes.25, 26, 27, 28 One result of this may be disruption of the phagosome membrane allowing M. tuberculosis to escape from its phagosome into the cytoplasm of mononuclear phagocytic cells.37 This phagosome rupture has itself been linked to the generation of cell death, possibly by the activation of NLRP3.38, 39, 40 ESAT-6 is however secreted in great quantity even in the extracellular environment. In fact, Cfp10 and ESAT-6 represent two of the most abundant proteins found in short-term culture supernatants of axenic broth culture.41,42 Thus, whether inside or outside of a host cell, secreted ESAT-6 has the opportunity to interact with both the internal and external membranes of the cell. This may be of particular relevance to development of the necrotic granuloma during which there is considerable neutrophil and monocyte death and M. tuberculosis is located both intra- and extracellularly.43
In support of ESAT-6 having an important extracellular function, we show in the present study and for the first time that exogenous ESAT-6 has a leukocidin-like action, inducing Ca2+ influx in aging neutrophils leading to calpain activation and necrosis. Thus ESAT-6 induced necrosis appears to occur by a similar mechanism to ionophore induced necrosis of neutrophils.24 Exactly how ESAT-6 induces Ca2+ movement across the cell membrane is unknown but previously others have suggested that it forms membrane pores.26 Indeed, pore formation is a common mechanism used by pathogenic bacteria to lyse host cells—for instance Escherichia coli haemolysins,44 Listeria listeriolysins45 and Streptococcus pyogenes streptolysins O and S.46 The ESAT-6/Cfp10 complex does not have the electrostatic surface properties typical of pore forming proteins and appears to bind to cell surface receptors.36 However, there is good evidence that ESAT-6 can dissociate from Cfp10 in certain conditions such as acidity,12 can undergo conformational change in the presence of phospholipids47 and can directly interact with membrane structure.25,28
It was striking that ESAT-6 specifically raised cytosolic Ca2+ levels and necrosis in phosphatidylserine externalised neutrophils with a negligible effect on cells without phosphatidylserine externalised. We hypothesise a number of explanations for this. Firstly, it is feasible that phosphatidylserine (PS) is a binding/interaction partner for ESAT-6 and thus membrane perturbation occurs only in PS externalised cells. Alternatively, the membrane repair mechanisms that confer resistance to bacterial and host derived membrane pores, such as endocytosis and lysosomal degradation of pore forming proteins48 may be less effective in PS externalised neutrophils. This is because effector proteins of these repair mechanisms including annexin A1 and synaptotagmin VII require PS to be available in the cytosolic leaflet of the cell membrane to catalyse their localisation to the cell membrane via their C2 domains. A similar mechanism may also explain the calpain dependence of necrosis that we observed, because calpain normally translocates to the cell surface upon activation in a process involving PS and elevated Ca2+.49 However, in cells with externalised PS there may be insufficient PS available in the cytosolic leaflet, thus the protease remains free to roam the cytosol in an uncontrolled but active proteolytic state resulting in necrosis.
We also used flow cytometry and confocal laser scanning microscopy to observe NET formation, demonstrating ESAT-6 treated necrotic neutrophils produced NETs. Induction of NETosis by pathogen derived membrane pores is a mechanism that has been identified recently, for example purified Panton–Valentine leukocidin (PVL) and leukotoxin GH from Staphylococcus aureus50 and leukotoxin from Mannheimia haemolytica caused NET formation.51 The mechanism of NETosis by bacterial toxins was previously unknown, but we provide evidence that, at least in the case of ESAT-6, it is Ca2+ dependent. This is supported by work from others that have showed calcium ionophore A2318752 and thapsigargin,53 which increases intracellular Ca2+ by inhibition of sarcoplasmic/endoplasmic ATPase (SERCA), can induce NET formation in a mechanism suggested to be through peptidylarginine deiminase (PAD) 4 hypercitrullination of histones.52,54 Indeed, we show that ESAT-6 and ionomycin treated neutrophils exhibited similar external localisation of MPO indicative of NET formation. NETosis has not been previously described as requiring PS externalisation.19 However, we have identified that, similar to secondary necrosis, ESAT-6 specifically induced NETosis in PS externalised cells.
Our observations that ESAT-6 causes Ca2+ dependent secondary necrosis or CAIN and NETosis provide a mechanistic basis for previous observations that virulent strains of M. tuberculosis carrying the ESX-1 secretion system can kill neutrophils when in co-culture.12 This leukocidin virulence mechanism may remove the antimycobacterial threat of the neutrophil while simultaneously generating nutrient rich necrotic tissue that fuels extracellular growth of the mycobacterium. This is consistent with observations of human tubercle lesions packed with dying neutrophils2 and more recent observations that the development of caseous, necrotic granulomas in a mouse model of progressive TB were associated with the presence of extracellular bacteria, neutrophil necrosis and NET-like structures.55 Thus ESAT-6 induction of NETs may be an important mechanism for the generation of lung pathology associated with active transmissible tuberculosis. There remains the possibility that other M. tuberculosis molecules can also induce neutrophil death because RD1 deleted mutants retain a residual cytotoxicity.12 Future work will aim to identify further mycobacterial effectors of neutrophil necrosis and establish if pharmacologic intervention of neutrophil necrosis alters pathology during infection.
Materials and Methods
Samples
Neutrophils were isolated from whole blood samples from healthy volunteers (BCG vaccinated) obtained through favourable ethical opinion by the University of Surrey Ethical Board.
Reagents
Dextran and ficoll (Sigma-Aldrich, Gillingham, Dorset, UK) were used for neutrophil purification from whole blood. Recombinant ESAT-6 expressed in Lactococcus lactis (Statum Serum Institute, Denmark) was used for experiments. Recombinant M. tuberculosis protein Rv0435c was used as a non-ESX-1 related protein control. Ionomycin was added to cells at 5 μM. Viability of neutrophils was measured using propidium iodide at 500 nM (Sigma-Aldrich). For flow cytometry, phosphatidylserine externalisation was detected using annexin V-FITC (2.5 μg/ml) and for confocal laser scanning microscopy annexin V-alexa fluor 647 (2.5 μg/ml) was used (BioLegend, San Diego, CA, USA). Fluo4-AM (10 μM) was used to measure Ca2+ concentration (Invitrogen Life Technologies, Paisley, UK). FITC conjugated antibodies to MPO and isotype control (347201—BioLegend), were used for flow cytometric analysis of neutrophil death.
Neutrophil preparation
Buffy coats were prepared by dextran sedimentation and haemolysis as previously described.53,54 For further purification, the resulting white blood cells were separated using a ficoll density gradient.56 The neutrophils were suspended in RPMI with 10% FCS, stable l-glutamine (0.29 mg/ml), penicillin (0.1 mg/ml), streptomycin (0.1 mg/ml), and FCS (10%) and maintained at 37 °C, 5% CO2 overnight, in cell culture flasks before treatments and further analysis. Before manipulation, neutrophil numbers were counted using a haemocytometer and purity determined (>95%) (NanoEnTek, Seoul, Korea).
Measurement of neutrophil necrosis
At the beginning of each experiment, the viability and phosphatidylserine externalisation of the aged neutrophils was measured using propidium iodide and annexin V-FITC, respectively, by flow cytometry using a FACS Canto (Becton Dickinson, Oxford, UK). ESAT-6 (20 or 40 μg/ml as stated), PBS vehicle or control protein was then added at the concentration stated in text. Proteins were added from a 2x stock solution to reduce locality artefacts. The increase in necrosis was then measured in each population as an increase in the percentage of propidium iodide positive neutrophils (as determined by gating). Where stated, the data was normalised to the increase in necrosis in vehicle only controls to enable comparisons between donors and expressed as a percentage increase.
Confocal laser scanning microscopy of neutrophils
Images were produced on either a LSM 510 (Zeiss, Oberkochen, Germany) or A1M (Nikon, Tokyo, Japan) CLSM with the neutrophils kept in glass-bottomed petri dishes (MatTek, Ashland, MA, USA). Measurements were obtained of the fluo4-AM dyed neutrophils by same sized region of interest tracking through the time-course of the experiment with the mean fluorescence intensity (MFI) measured. This was divided by the MFI of time 0 (F/F0), or the closest recording to that point. To determine whether the neutrophils had externalised phosphatidylserine, they were also stained prior to stimulation with annexinV-fluor647. To confirm necrosis, we stained for extracellular DNA with propidium iodide. Images were analysed uniformly between controls and treatment using NIS Elements (Nikon) or Zen 2009 (Zeiss), and images produced using ImageJ (NIH).
Localisation of myeloperoxidase in Neutrophil Extracellular Traps
We examined the extruded MPO content of NETs stimulated by the addition of ESAT-6 at 40 μg/ml for 1 h or ionomycin at 5 μM for 30 min. The cells were centrifuged at 1000rcf for 1 min to create a pellet. The media was removed and the neutrophils were resuspended in ‘blocking buffer’ (5% heat inactivated normal human serum and 1% bovine serum albumin in PBS). The FITC-conjugated antibodies for MPO or the isotype control were added in PBS with block at 1 : 50 for 30 min. The neutrophils were centrifuged and resuspended in PBS ready for flow cytometric analysis or CLSM imaging.
Statistical analysis
The results are presented as the mean+S.E.M. All statistics are Student’s t test or ANOVA with Dunnett’s post test as stated.
Abbreviations
- ANOVA:
-
analysis of variance
- AnV:
-
annexin V
- BCG:
-
Bacille Calmette Guérin
- Ca2+:
-
calcium
- CAIN:
-
Ca2+ induced necrosis
- Cfp10:
-
culture filtrate protein 10 kDa
- CLSM:
-
confocal laser scanning microscopy
- DMSO:
-
dimethyl sulfoxide
- ESAT-6:
-
6 kDa early secretory antigenic target
- ESX-1:
-
ESAT-6 secretion system-1
- FCS:
-
foetal calf serum
- FITC:
-
fluorescein isothiocyanate
- MFI:
-
mean fluorescence intensity
- MPO:
-
myeloperoxidase
- NET:
-
neutrophil extracellular trap
- NLRP3:
-
NOD-like receptor family, pyrin domain containing 3
- PI:
-
propidium iodide
- PS:
-
phosphatidylserine
- RD1:
-
Region of Difference 1
- ROS:
-
reactive oxygen species
- S.E.M.:
-
standard error of the mean
- TB:
-
tuberculosis
References
W.H.O. Report. World Health Organisation GLOBAL TUBERCULOSIS REPORT 2013.
Medlar EM . A study of the process of caseation in tuberculosis. Am J Pathol 1926; 2 275–U278.
Butler RE, Brodin P, Jang J, Jang MS, Robertson BD, Gicquel B et al. The balance of apoptotic and necrotic cell death in Mycobacterium tuberculosis infected macrophages is not dependent on bacterial virulence. PLoS One 2012; 7: e47573.
Hinchey J, Lee S, Jeon BY, Basaraba RJ, Venkataswamy MM, Chen B et al. Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J Clin Invest 2007; 117: 2279–2288.
Velmurugan K, Chen B, Miller JL, Azogue S, Gurses S, Hsu T et al. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. Plos Pathog 2007; 3: 972–980.
Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM . Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat Immunol 2010; 11: 751–758.
Keane J, Remold HG, Kornfeld H . Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol 2000; 164: 2016–2020.
Russell DG, Barry 3rd CE, Flynn JL . Tuberculosis: what we don't know can, and does, hurt us. Science 2010; 328: 852–856.
Lee J, Remold HG, Ieong MH, Kornfeld H . Macrophage apoptosis in response to high intracellular burden of Mycobacterium tuberculosis is mediated by a novel caspase-independent pathway. J Immunol 2006; 176: 4267–4274.
Jones GS, Amirault HJ, Andersen BR . Killing of Mycobacterium tuberculosis by neutrophils - a nonoxidative process. J Infect Dis 1990; 162: 700–704.
Lowe DM, Redford PS, Wilkinson RJ, O'Garra A, Martineau AR . Neutrophils in tuberculosis: friend or foe? Trends Immunol 2012; 33: 14–25.
Corleis B, Korbel D, Wilson R, Bylund J, Chee R, Schaible UE . Escape of Mycobacterium tuberculosis from oxidative killing by neutrophils. Cell Microbiol 2012; 14: 1109–1121.
Yang CT, Cambier CJ, Davis JM, Hall CJ, Crosier PS, Ramakrishnan L . Neutrophils exert protection in the early Tuberculous granuloma by oxidative killing of mycobacteria phagocytosed from infected macrophages. Cell Host Microbe 2012; 12: 301–312.
Briken V . "With a little Help from my friends": efferocytosis as an antimicrobial mechanism. Cell Host Microbe 2012; 12: 261–263.
Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence (vol 393, p 537, 1998). Nature 1998; 396: 190–198.
Mahairas GG, Sabo PJ, Hickey MJ, Singh DC, Stover CK . Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M-bovis. J Bacteriol 1996; 178: 1274–1282.
Sorensen AL, Nagai S, Houen G, Andersen P, Andersen AB . Purification and characterization of a low-molecular-mass T-cell antigen secreted by Mycobacterium tuberculosis. Infect Immun 1995; 63: 1710–1717.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS et al. Neutrophil extracellular traps kill bacteria. Science 2004; 303: 1532–1535.
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 2007; 176: 231–241.
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 2007; 13: 463–469.
Brinkmann V, Zychlinsky A . Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol 2007; 5: 577–582.
Brinkmann V, Zychlinsky A . Neutrophil extracellular traps: is immunity the second function of chromatin?. J Cell Biol 2012; 198: 773–783.
Ramos-Kichik V, Mondragon-Flores R, Mondragon-Castelan M, Gonzalez-Pozos S, Muniz-Hernandez S, Rojas-Espinosa O et al. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis 2009; 89: 29–37.
Francis RJ, Kotecha S, Hallett MB . Ca2+ activation of cytosolic calpain induces the transition from apoptosis to necrosis in neutrophils with externalized phosphatidylserine. J Leukoc Biol 2013; 93: 95–100.
Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM et al. The primary mechanism of attenuation of Bacillus Calmette–Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci USA 2003; 100: 12420–12425.
Smith J, Manoranjan J, Pan M, Bohsali A, Xu JJ, Liu J et al. Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect Immun 2008; 76: 5478–5487.
De Leon J, Jiang GZ, Ma Y, Rubin E, Fortune S, Sun JJ . Mycobacterium tuberculosis ESAT-6 exhibits a unique membrane-interacting activity that is not found in its ortholog from non-pathogenic Mycobacterium smegmatis. J Biol Chem 2012; 287: 44184–44191.
Jonge MI, Pehau-Arnaudet G, Fretz MM, Romain F, Bottai D, Brodin P et al. ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. J Bacteriol 2007; 189: 6028–6034.
Yang R, Xi C, Sita DR, Sakai S, Tsuchiya K, Hara H et al. The RD1 locus in the Mycobacterium tuberculosis genome contributes to the maturation and secretion of IL-1alpha from infected macrophages through the elevation of cytoplasmic calcium levels and calpain activation. Pathog Dis 2014; 70: 51–60.
Kinhikar AG, Verma I, Chandra D, Singh KK, Weldingh K, Andersen P et al. Potential role for ESAT6 in dissemination of M. tuberculosis via human lung epithelial cells. Mol Microbiol 2010; 75: 92–106.
Macdonald SHF, Woodward E, Coleman MM, Dorris ER, Nadarajan P, Chew WM et al. Networked T Cell Death following Macrophage Infection by Mycobacterium tuberculosis. PLoS One 2012; 7: 6.
Paredes RM, Etzler JC, Watts LT, Zheng W, Lechleiter JD . Chemical calcium indicators. Methods 2008; 46: 143–151.
Wang KKW, Nath R, Posner A, Raser KJ, BurokerKilgore M, Hajimohammadreza I et al. An alpha-mercaptoacrylic acid derivative is a selective nonpeptide cell-permeable calpain inhibitor and is neuroprotective. P Natl Acad Sci USA 1996; 93: 6687–6692.
Parker H, Albrett AM, Kettle AJ, Winterbourn CC . Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukoc Biol 2012; 91: 369–376.
Simeone R, Bottai D, Brosch R . ESX/type VII secretion systems and their role in host–pathogen interaction. Curr Opin Microbiol 2009; 12: 4–10.
Renshaw PS, Lightbody KL, Veverka V, Muskett FW, Kelly G, Frenkiel TA et al. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J 2005; 24: 2491–2498.
Houben D, Demangel C, van Ingen J, Perez J, Baldeon L, Abdallah AM et al. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol 2012; 14: 1287–1298.
Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R et al. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. Plos Pathog 2012; 8: e1002507.
Wong KW, Jacobs WR Jr . Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cell Microbiol 2011; 13: 1371–1384.
Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF et al. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol 2010; 12: 1046–1063.
Lange S, Rosenkrands I, Stein R, Andersen P, Kaufmann SH, Jungblut PR . Analysis of protein species differentiation among mycobacterial low-Mr-secreted proteins by narrow pH range Immobiline gel 2-DE-MALDI-MS. J Proteomics 2014; 97: 235–244.
Mattow J, Jungblut PR, Schaible UE, Mollenkopf HJ, Lamer S, Zimny-Arndt U et al. Identification of proteins from Mycobacterium tuberculosis missing in attenuated Mycobacterium bovis BCG strains. Electrophoresis 2001; 22: 2936–2946.
Orme IM . A new unifying theory of the pathogenesis of tuberculosis. Tuberculosis (Edinb) 2014; 94: 8–14.
Cavalieri SJ, Snyder IS . Effect of Escherichia coli alpha-hemolysin on human peripheral leukocyte viability in vitro. Infect Immun 1982; 36: 455–461.
Gekara NO, Westphal K, Ma B, Rohde M, Groebe L, Weiss S . The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Cell Microbiol 2007; 9: 2008–2021.
Sierig G, Cywes C, Wessels MR, Ashbaugh CD . Cytotoxic effects of streptolysin o and streptolysin s enhance the virulence of poorly encapsulated group a streptococci. Infect Immun 2003; 71: 446–455.
Meher AK, Bal NC, Chary KV, Arora A . Mycobacterium tuberculosis H37Rv ESAT-6-CFP-10 complex formation confers thermodynamic and biochemical stability. FEBS J 2006; 273: 1445–1462.
Cassidy SK, O'Riordan MX . More than a pore: the cellular response to cholesterol-dependent cytolysins. Toxins 2013; 5: 618–636.
Benyamin Y . The structural basis of calpain behavior. Febs J 2006; 273: 3413–3414.
Malachowa N, Kobayashi SD, Freedman B, Dorward DW, DeLeo FR . Staphylococcus aureus leukotoxin GH promotes formation of neutrophil extracellular traps. J Immunol 2013; 191: 6022–6029.
Aulik NA, Hellenbrand KM, Klos H, Czuprynski CJ . Mannheimia haemolytica and its leukotoxin cause neutrophil extracellular trap formation by bovine neutrophils. Infect Immun 2010; 78: 4454–4466.
Wang Y, Li M, Stadler S, Correll S, Li P, Wang D et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 2009; 184: 205–213.
Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. Febs Lett 2010; 584: 3193–3197.
Neeli I, Khan SN, Radic M . Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol 2008; 180: 1895–1902.
Marzo E, Vilaplana C, Tapia G, Diaz J, Garcia V, Cardona PJ . Damaging role of neutrophilic infiltration in a mouse model of progressive tuberculosis. Tuberculosis (Edinb) 2014; 94: 55–64.
Destin KG, Linden JR, Laforce-Nesbitt SS, Bliss JM . Oxidative burst and phagocytosis of neonatal neutrophils confronting Candida albicans and Candida parapsilosis. Early Hum Dev 2009; 85: 531–535.
Acknowledgements
We gratefully thank all of the donors that provided the blood samples, and to Dr J. Noel Wardell for taking them. This work was conducted in part using the University of Surrey Bioimaging and Flow Cytometry Core Facility. Finally we thank all those in the department of microbial and cellular sciences that provided insight and discussion. This work was supported by the Wellcome Trust grant WT090242MA and Rosetrees Trust grant A549.
Author Contributions
RJF and REB performed the experiments and subsequent analysis. RJF, GS and REB wrote the manuscript. RJF and GS conceived the idea and GS supervised the project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International Licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0
About this article

Cite this article
Francis, R., Butler, R. & Stewart, G. Mycobacterium tuberculosis ESAT-6 is a leukocidin causing Ca2+ influx, necrosis and neutrophil extracellular trap formation. Cell Death Dis 5, e1474 (2014). https://doi.org/10.1038/cddis.2014.394
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.394
This article is cited by
-
Effects of functionally diverse calpain system on immune cells
Immunologic Research (2021)
-
Fibrinogen protects neutrophils from the cytotoxic effects of histones and delays neutrophil extracellular trap formation induced by ionomycin
Scientific Reports (2020)
-
Mycobacterium bovis uses the ESX-1 Type VII secretion system to escape predation by the soil-dwelling amoeba Dictyostelium discoideum
The ISME Journal (2020)
-
Mycobacterium fortuitum-induced ER-Mitochondrial calcium dynamics promotes calpain/caspase-12/caspase-9 mediated apoptosis in fish macrophages
Cell Death Discovery (2018)
-
Complex regulation of neutrophil-derived MMP-9 secretion in central nervous system tuberculosis
Journal of Neuroinflammation (2017)
