
Abstract
p53 has a crucial role in governing cellular mechanisms in response to a broad range of genotoxic stresses. During DNA damage, p53 can either promote cell survival by activating senescence or cell-cycle arrest and DNA repair to maintain genomic integrity for cell survival or direct cells to undergo apoptosis to eliminate extensively damaged cells. The ability of p53 to execute these two opposing cell fates depends on distinct signaling pathways downstream of p53. In this study, we showed that under DNA damage conditions induced by chemotherapeutic drugs, gamma irradiation and hydrogen peroxide, p53 upregulates a novel protein, proline-rich acidic protein 1 (PRAP1). We identified functional p53-response elements within intron 1 of PRAP1 gene and showed that these regions interact directly with p53 using ChIP assays, indicating that PRAP1 is a novel p53 target gene. The induction of PRAP1 expression by p53 may promote resistance of cancer cells to chemotherapeutic drugs such as 5-fluorouracil (5-FU), as knockdown of PRAP1 increases apoptosis in cancer cells after 5-FU treatment. PRAP1 appears to protect cells from apoptosis by inducing cell-cycle arrest, suggesting that the induction of PRAP1 expression by p53 in response to DNA-damaging agents contributes to cancer cell survival. Our findings provide a greater insight into the mechanisms underlying the pro-survival role of p53 in response to cytotoxic treatments.
Similar content being viewed by others
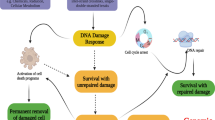


Main
Maintenance of genomic stability is essential for the accurate duplication and transmission of genetic material during cell division so as to ensure the survival of organisms. Failure to repair DNA damage results in diverse disorders and contributes to cancer development.1 In response to DNA damage, cells activate surveillance mechanisms, such as DNA repair, cell-cycle checkpoint arrest and apoptosis.
The tumor-suppressor gene, p53, has a critical role in regulating the responses of mammalian cells to DNA damage. Under normal circumstances, p53 is present in low amounts with a short half-life. p53 levels are tightly regulated by an oncoprotein, Mdm2, which promotes the ubiquitination and rapid degradation of p53 when it binds to the N-terminus of the p53 protein.2 However, upon DNA damage, Mdm2 is rapidly degraded, leading to accumulation of p53 as a result of the disrupted p53–Mdm2 interaction. The transcriptional activity of p53 is then activated by a series of modulators such as the kinases ATM and ATR, as well as acetylases such as p300/CBP and p300/CBP-associated factor.3 The transcriptional targets of p53 regulate various mechanisms involved in progression through the cell cycle. In response to DNA damage, p53 inhibits the entry of cells into the DNA synthesis phase of the cell cycle and activates DNA repair mechanisms.4 Alternatively, extensively damaged cells are eliminated through apoptosis.5 However, the mechanisms leading to the determination of cell fate resulting in either cell survival or death are still unclear.
p53 is a well-known guardian of genome stability and one of the most commonly inactivated genes in human cancers.6 The loss of p53 function predisposes one to cancer development because DNA damage resulting from cellular stresses, such as ultraviolet radiation and carcinogens, may not be repaired or removed by apoptosis but accumulates in subsequent generations of cells. The role of p53 in the response of cancer cells to chemotherapy is not entirely clear. On the one hand, p53 may enhance the recovery of cancer cells by activating DNA repair pathways,7 while on the other, it regulates apoptosis and has been shown to increase sensitivity of cells to chemotherapeutic agents.8 The apparent discrepancy may be explained by genes targeted by p53 under different circumstances, resulting in different cell fates. A characterization of new p53 responsive genes is hence required for a better understanding of how p53 determines cell fates.
In this study we show that PRAP1, a novel gene encoding proline-rich acidic protein 1 (PRAP1), is a p53-responsive gene induced by genotoxic stress. PRAP1 was originally identified as a differentially expressed gene in the pregnant uterus of rodents.9 We have previously cloned the human PRAP1 gene and showed that PRAP1 is expressed in the epithelium of the liver, gastrointestinal tract and kidneys.10 Although the functions of PRAP1 are unknown, our previous studies suggest that it inhibits tumor cell growth. Here, we report that PRAP1 expression is induced in a p53-dependent manner when cells are treated with DNA-damaging agents. We show that the PRAP1 gene is a novel transcriptional target of p53 and contains functional p53-response elements. Furthermore, the reduction of PRAP1 expression by small-interfering RNA (siRNA) knockdown increases the sensitivity of cells to DNA-damaging agents such as 5-fluorouracil (5-FU), with enhanced DNA damage and caspase-dependent apoptosis. PRAP1 possibly protects cells from apoptosis partly through the induction of cell-cycle arrest, as we observed a significant attenuation of 5-FU-induced cell-cycle arrest in PRAP1 knockdown cells. Our data indicate that PRAP1 modulates cell fate downstream of p53 activation by DNA-damaging agents and suggest that PRAP1 may be an important determinant of cancer cell survival.
Results
PRAP1 expression is induced by DNA-damaging agents
The regulation of PRAP1 expression by cellular stresses was studied using the human colon cancer cell line, HCT116. Cells were exposed to nucleic acid-damaging agents such as 5-FU, camptothecin (CPT) and etoposide as well as nutrient deprivation, ethanol and hydrogen peroxide. The expression of PRAP1 mRNA was highly induced by the chemotherapeutic agents at 48 h of treatment and, to a lesser degree, by hydrogen peroxide (Figure 1a). There was no detectable induction of PRAP1 mRNA expression by serum deprivation and ethanol. As these chemotherapeutic agents and hydrogen peroxide are well documented for their DNA-damaging effects,11, 12, 13 we surmised that PRAP1 is likely to be a genotoxic responsive gene. The induction of PRAP1 by DNA-damaging agents was also observed in HepG2, a liver cancer cell line, suggesting that this induction is not cell line-specific (Figure 1b).

PRAP1 is induced by DNA-damaging agents. (a) PRAP1 mRNA is induced by DNA-damaging agents in HCT116 cells. Reverse transcriptase (RT)-PCR gel picture showing the expression of prap1 and gapdh (glyceraldehyde 3-phosphate dehydrogenase) in HCT116 cells after treatment with the indicated stressors 5-FU (25 μM), CPT (20 nM), etoposide (20 μM), 5% ethanol or serum-free medium for 48 h; hydrogen peroxide (100 μM) for 1 h and recovered for 4 h. With the exception of ethanol and serum deprivation, PRAP1 mRNA expression was induced by all the other agents. (b) PRAP1 mRNA is induced by DNA-damaging agents in HepG2 cells. RT-PCR gel picture showing the increased expression of prap1 and gapdh in HepG2 cells after treatment with 5-FU (25 μM) and CPT (20 nM) for 48 h. (c) PRAP1 mRNA is induced by DNA-damaging agents in a dose-dependent manner. RT-PCR gel picture showing the enhanced expression of prap1 and gapdh in HCT116 cells after treatment with 5-FU and CPT at the indicated doses for 24, 48 and 72 h. (d) PRAP1 protein is induced by DNA-damaging agents in a time-dependent manner. Western blot showing the expression of PRAP1 and GAPDH in HCT116 cells after treatment with 5-FU (25 μM) and CPT (20 nM) for 24, 48 and 72 h. (e) PRAP1 and p21 mRNA are induced by gamma irradiation. Graph shows fold induction of prap1 and p21 expression in HCT116 cells after treatment with gamma irradiation at 2, 5 and 10 Gy and recovered at 1, 2, 4 and 8 h. Real-time RT-PCR was performed and relative expression of prap1 and p21 were normalized against gapdh and calculated as fold induction
5-FU and CPT are drugs commonly used in colorectal cancer chemotherapy.14 To ascertain the effects of these drugs on PRAP1 mRNA and protein levels, HCT116 cells were treated with different doses of 5-FU and CPT for up to 72 h. PRAP1 mRNA expression was induced in a dose-dependent manner by 24 h of treatment with either 5-FU or CPT (Figure 1c). PRAP1 mRNA expression was sustained longer in response to 5-FU (at least 72 h) compared with CPT. Increased levels of PRAP1 protein in response to 25 μM 5-FU and 20 nM CPT were detected from 24 h of treatment up to 72 and 48 h, respectively (Figure 1d). The temporal regulation of PRAP1 expression in response to 5-FU and CPT suggests that PRAP1 may function at specific molecular events downstream of pathways activated by genotoxic stress.
We further investigated PRAP1 expression in response to gamma irradiation, a well-characterized model to study DNA damage.15 Using quantitative real-time PCR, we showed that PRAP1 mRNA levels were induced by gamma irradiation in a dose- and time-dependent manner (Figure 1e). Ten gray (Gy) of gamma irradiation induced PRAP1 mRNA expression within 1 h after exposure and more than threefold increase following 2 h of exposure. An increased mRNA expression of p21, a known direct transcriptional target of p53 induced by genotoxic stress,16 was also observed within 1 h after exposure to 10 Gy of gamma irradiation. These results indicate that PRAP1 expression is induced under a variety of conditions that cause DNA damage.
Induction of PRAP1 expression by DNA-damaging agents is dependent on p53
As p53 has an important role in the response to DNA damage, we investigated whether induction of PRAP1 expression upon exposure to the DNA-damaging agents, 5-FU and CPT, is p53-dependent. A p53 knockout (p53−/−) HCT116 cell line was used. PRAP1 mRNA was induced in wild-type HCT116, but not in p53−/− cells, at both low and high doses of 5-FU and CPT (Figure 2a, top panel), suggesting that p53 is necessary for the induction of PRAP1 expression by DNA-damaging agents.

Induction of PRAP1 by DNA-damaging agents is dependent on p53. (a, top panel) Cells without p53 failed to induce PRAP1 when treated with DNA-damaging agents. Reverse transcriptase (RT)-PCR gel picture showing the expression of prap1 and gapdh (glyceraldehyde 3-phosphate dehydrogenase) in HCT116 cells and significantly reduced expression of prap1in the p53-nulll HCT116 derivative, p53−/−, after treatment with the indicated doses of 5-FU and CPT for 24 h. (a, bottom panel) Reintroduction of wild-type p53 into p53−/− cells restored PRAP1 induction by DNA-damaging agents. RT-PCR gel picture showing the expression of prap1 and gapdh in p53−/− cells after transfection with either empty vector control (V) or wild-type p53 (WT) or mutant p53 (Mut), followed by treatment with either 5-FU (25 μM) or CPT (20 nM) for 24 h. Transfection with WT restored PRAP1 induction by 5-FU and CPT. (b, top panel) Western blot picture showing p53 protein was efficiently knocked down by siRNA in both HCT116 and RKO cells. (b, bottom panel) Both p53-depleted cell lines failed to induce PRAP1 upon 5-FU treatment. Graphs showing induction of prap1 expression in HCT116 and RKO cells after treatment with 5-FU (2.5 or 25 μM) for 24 h. Real-time RT-PCR was performed and relative expression of prap1 were normalized against gapdh and calculated as fold induction. p53 knockdown cells showed marked reduction in the induction of PRAP1 expression after 5-FU treatment as compared with the respectively control siRNA-treated cells (*P<0.05). (c) Schematic diagram of p53 binding sites in PRAP1 gene construct. Two p53 binding sites were identified in intron 1 of the PRAP1 gene, starting at +1316 and +1460, while the transcription start site of PRAP1 gene is denoted as +1. (d, left panel) The two p53-response elements (blue) within the first intron 1 of PRAP1 gene were cloned into the luciferase reporter plasmid (pGL3-Promoter) upstream of the SV 40 promoter to generate the pGL3-PRAP plasmid. (d, right panel) Figure showing the fold induction of SV40 promoter activity of pGL3-P, pGL3-PRAP and pGL3-p21 cotransfected with pcDNA vector, pcDNA-p53 and pcDNA-p53Mut in p53−/− cells for 24 h. For each transfection, the Firefly luciferase activity was normalized with the Renilla reniformis luciferase activity by the cotransfected pRL-TK (thymidine kinase promoter-Renilla luciferase reporter plasmid). The relative activity of each construct is compared against the activity of the pGL3-P. pGL3-P, basic luciferase promoter; pGL3-PRAP, basic luciferase promoter plasmid constructed with the two p53 binding elements of PRAP1 gene; pGL3-p21, basic luciferase promoter plasmid constructed with the p53 binding elements of p21 gene (serving as a positive control for comparison); Vector: pcDNA vector; p53 WT: pcDNA with wild-type p53 construct; p53 Mut: pcDNA with mutant p53. (e, top panel) Schematic diagram of chromatin immunoprecipitation (ChIP) primers used to amplify the two p53-response elements and non-specific regions in intron 1 of PRAP1 gene. (e, middle panel) p53 directly interacted with the two p53-response elements in PRAP1 gene. HCT116 cells were treated with 5-FU (25 or 50 μM) for 24 h. The chromatin bound with p53 were isolated and amplified by PCR. A primer pair recognizing the known p53-response element on p21 promoter was used as a positive control. (e, bottom panel) p53 specifically bound to the p53-response elements (P1, P2). Primer pairs that align to non-specific regions in intron 1 did not amplify a band (N1, N2 and N3). The ChIP-precipitated DNA and input DNA were from cells treated with 25 μM 5-FU
To investigate whether the induction of PRAP1 mRNA expression is dependent on the transcriptional activity of p53, we reintroduced into p53−/− cells either the wild-type p53 by transfecting with pCMV-p53 (WT) or the DNA-binding deficient mutant p53 construct pCMV-p53mt153 (Mut). Transfection of wild-type p53 into p53−/− cells rescued the induction of PRAP1 mRNA by 5-FU and CPT (Figure 2a, bottom panel). However, PRAP1 mRNA could not be induced in mutant or empty vector (V)-transfected p53−/− cells. This indicated that the induction of PRAP1 expression by DNA-damaging agents requires the presence of wild-type p53 with intact DNA-binding activity.
The dependence of PRAP1 induction on p53 was further confirmed by siRNA knockdown of p53 in two colorectal cancer cell lines that contain wild-type p53, HCT116 and RKO. The induction of PRAP1 mRNA was abrogated upon DNA damage induced by 5-FU when the endogenous p53 was depleted by siRNA in these cell lines (Figure 2b, middle and bottom panels). Our data indicates that PRAP1 is a likely novel transcriptional target gene of p53.
PRAP1 gene contains functional p53-response elements
As PRAP1 is a possible target of p53 transcriptional activity, we investigated whether the PRAP1 gene contains p53-response elements. Using the p53MH algorithm designed to search for potential p53 binding sites,17 we identified two putative p53-response elements within the first intron of PRAP1. The sequences of these two p53-response elements in PRAP1 matched the consensus p53 binding site by 84% and 70%, while the known p53-response element in the p21 promoter showed a match of 90% (Table 1). The human PRAP1 gene comprises five exons10 and the positions of the two putative p53-response elements (starting at +1316 and +1460, respectively) in PRAP1 are illustrated (Figure 2c).
To determine whether the two identified p53-response elements in PRAP1 (PRAP1-p53BS) are functional, we amplified the intronic fragment from positions +1197 to +1534, which spans across both putative p53-response elements of PRAP1. This fragment was cloned into a pGL3-Promoter luciferase reporter vector upstream of a minimal SV-40 promoter, forming the pGL3-PRAP1 construct (Figure 2d, left panel). To examine the responsiveness of these two potential p53-response elements in intron 1 of PRAP1, the pGL3-PRAP1 construct was transiently cotransfected with empty pCMV (Vector), pCMV-p53 (p53 WT) or pCMV-p53mt153 (p53 Mut) into p53−/− cells. The p53-response element in p21 (pGL3-p21) was used as a positive control in the co-transfection experiments as p21 is reported to be a direct transcriptional target of p53,16 while the empty pGL3-Promoter vector was used as a negative control. We found that co-transfection of p53 WT, but not Vector or p53 Mut, increased the SV-40 promoter activities by about threefold in cells transfected with pGL3-PRAP1 (Figure 2d, right panel). These results confirm that the predicted p53-response elements in PRAP1 are responsive to the transcriptional activity of p53.
Chromatin immunoprecipitation (ChIP) studies were performed to determine the physical binding of p53 to these two identified response elements. Two pairs of ChIP primers (ChIP P1 covering +1255 to 1362; ChIP P2 covering +1396 to 1503) were designed to amplify the two p53-response elements (from +1316 to 1349 and +1460 to 1482) in intron 1 of PRAP1 gene, respectively (illustrated in Figure 2e, top panel). In HCT116 cells treated with 5-FU, there were dose-dependent increases in the binding of p53 to both the response elements in PRAP1 gene (Figure 2e, middle panel). This indicates that the p53 protein interacts directly with the two novel p53-response elements in PRAP1 gene and confirms that PRAP1 is a novel target gene of p53 under DNA damage conditions. Furthermore, we showed that unlike P1 and P2 that align to the p53-response element, the primers (N1, N2 and N3) designed to recognize regions upstream and downstream from the p53-response element failed to generate a PCR product (Figure 2e, bottom panel).
Knockdown of PRAP1 enhances DNA damage by genotoxic agents and reduces cell survival
To investigate the biological functions of p53-mediated PRAP1 induction, we determined the effects of PRAP1 siRNA knockdown on p53-dependent cellular responses, such as DNA repair and apoptosis. To account for non-specific effects, control siRNAs containing mutations in four bases of their corresponding PRAP1 siRNAs were used, which do not target PRAP1 and other genes. The 5-FU-induced PRAP1 mRNA and protein expression levels were effectively repressed by both PRAP1 siRNAs (Figures 3a and b) as compared with their respective control siRNAs.

Knockdown of PRAP1 sensitizes cells to DNA-damaging agents. HCT116 cells were transfected with PRAP1-specific siRNAs (PRAP1 siRNA1 and 2) or PRAP1 siRNA specific control siRNAs (control siRNA1 and 2) for 48 h and followed by the addition of 5-FU for another 24 h. PRAP1 knockdown significantly reduced 5-FU-induced PRAP1 mRNA and protein expression. (a and b) PRAP1 knockdown by siRNA reduces PRAP1 mRNA and protin expression induced by 5-FU treatment. (c) Knockdown of PRAP1 expression enhances 5-FU-induced DNA damage. PRAP1 knockdown increased the tail movement of 5-FU-treated cells (left panel), as well as the number of micronuclei (MN) formation (right panel), indicating enhanced DNA damage in these cells. (d) Knockdown of PRAP1 expression reduces colony numbers after 5-FU treatment. HCT116 cells were treated as described above with 3 μM or 6 μM 5-FU before re-culturing for colony formation assay. Colonies were stained with crystal violet for visualization and analysis using ImageJ. PRAP1 knockdown reduced the number of colonies formed by cells treated with both doses of 5-FU. (*P<0.05). GAPDH, glyceraldehyde 3-phosphate dehydrogenase
We examined the extent of 5-FU-induced DNA damage in the presence or absence of PRAP1. PRAP1 knockdown enhanced the DNA damage induced by 5-FU by 2.5-fold, as evidenced by the increase in tail movement in the comet assay (Figure 3c, left panel). Furthermore, PRAP1 knockdown resulted in a marked increase in micronuclei frequency in cells, a reliable indicator of chromosomal damage and genomic instability18 (4.3% versus 0.74%, Figure 3c, right panel). Our findings suggest that PRAP1 normally functions to minimize genomic insults in cells.
The effect of PRAP1 on cell survival was studied in the presence of 5-FU in HCT116 cells using the in vitro colony formation assay. In the 5-FU-treated HCT116 cells, PRAP1 knockdown significantly reduced the number and size of colonies formed compared with the control siRNA treatment (Figure 3d, top panel). There was about 50% reduction in the number of colonies formed by PRAP1 knockdown cells treated with 3 μM 5-FU, and 80% reduction at a higher dose of 6 μM 5-FU (Figure 3d, bottom figure). This indicates that the stress-induced PRAP1 response confers survival advantage to cells and suggests that loss of PRAP1 may impair DNA repair and facilitate cell death.
PRAP1 is crucial for S-phase arrest induced by 5-FU, a DNA-damaging agent
Upon DNA damage, mammalian cells may activate cell-cycle arrest via the p53 pathway to repair the damage.19 As the reduction in PRAP1 expression is associated with increased DNA damage, we postulate that PRAP1 may protect cells by regulating cell-cycle progression and/or DNA repair pathways. We examined the effects of 5-FU treatment on the cell-cycle profiles of HCT116 cells in the presence or absence of PRAP1 using 5-bromo-2′deoxyuridine/7-amino-actinomycin D (BrdU/7-AAD) analysis. Cells were synchronized in serum-free media for 18 h before 24 h of 5-FU treatment, followed by pulsed labeling with BrdU for 30 min. To investigate cell death, we performed propidium iodide staining of fixed cells to ascertain the percentage of cells in the sub-G1 population.
Our results indicate that the 24-h treatment of control siRNA-transfected cells with 5-FU primarily resulted in cell-cycle arrest at S-phase (Figure 4a), with minimal cell death (Figure 4b). Knockdown of PRAP1 by siRNA attenuated the 5-FU-induced S-phase arrest by at least 51% (Figure 4a), with a significant increase in sub-G1 phase indicating cell death (Figure 4b). In the absence of 5-FU, no significant differences in cell-cycle profiles were observed in PRAP1 siRNA knockdown cells compared with control siRNA-transfected cells (Figure 4a, bottom panel). Pre-treatment of cells with the pan-caspase inhibitor, Z-VAD, effected minimal S-phase recovery in PRAP1 knockdown cells, based on the propidium iodide flow cytometry analysis (Figure 4c). This suggests that the attenuation of S-phase arrest in PRAP1-depleted cells treated with 5-FU was not due to apoptosis of S-phase cells.

S-phase arrest induced by 5-FU is abrogated by PRAP1 knockdown. (a) Cells were serum starved for 18 h followed by 24 h of 5-FU treatment in media containing 10% fetal bovine serum (FBS). Control cells were treated with 10% FBS in parallel. The distribution of cell-cycle phases of HCT116 was determined by flow cytometry after staining the cells with BrdU and 7-AAD. The populations of cells at G0/G1, S- and G2/M phases are indicated in the boxes. (b) Significant increases in sub-G1 population measured by propidium iodide (PI) staining were observed in cells transfected with PRAP1 siRNAs compared with control siRNA-transfected and untransfected cells. (c) Inhibition of apoptosis by pan caspase inhibitor, Z-VAD, did not rescue the loss of S-phase arrest in PRAP1-depleted cells. Z-VAD was added 1 h before 5-FU treatment in PRAP1 knockdown cells and the percentage of S-phase population were determined by PI flow cytometry analysis. (d) The abrogation in S-phase was accompanied by a reduction in S-phase cyclin proteins, cyclin A and CDK2. Western blot shows the expression of cyclin E, cyclin A, CDK4, CDK2, p53 and B-actin (loading control) in HCT116 cells after transfection with PRAP1 siRNA or control siRNA for 48 h and followed by the addition of 5-FU for 24 h
We examined the levels of G1/S phase regulatory proteins, including cyclins A and E, as well as cyclin-dependent kinases (CDKs), including CDK2 and CDK4. Consistent with other studies,20, 21 we found that 5-FU treatment induced increases in the expression of p53 and S-phase proteins, cyclin E and cyclin A (Figure 4d). PRAP1 knockdown by siRNA markedly reduced cyclin A expression in 5-FU-treated cells and slightly decreased cyclin E level, whereas p53 expression level remained unchanged. There was also a reduction in CDK2 but not CDK4 levels. Therefore, the protective mechanism conferred by PRAP1 against DNA-damaging agents through the induction of S-phase arrest may be mediated by the effect of PRAP1 on S-phase cyclins and CDK2. As cell-cycle arrest is potentially a cell-survival strategy, the failure of PRAP1 knockdown cells to undergo arrest may explain our earlier observations that loss of PRAP1 sensitizes cells to 5-FU. Taken together, our observations highlight the importance of PRAP1 in mediating S-phase arrest and cell survival during DNA damage conditions.
We were also curious to find out whether PRAP1 is required for S-phase arrest under other cellular stresses which do not induce DNA damage, such as thymidine block. PRAP1 mRNA expression was highly upregulated during the S-phase arrest induced by thymidine block (Supplementary Figure S1A), consistent with other studies.22, 23 Thus PRAP1 expression may be induced by non-DNA-damaging cellular stresses, possibly to impede cell-cycle progression. No PRAP1 induction was observed during G2/M phase arrest induced by taxol or nocodazole. Interestingly, thymidine also induced the expression of p53 in both HCT116 and RKO cells (Supplementary Figure S1B), in agreement with previous observations on cellular responses to thymidine.24 The induction of both p53 and PRAP1 by thymidine treatment is consistent with our observations that PRAP1 is a p53-target gene. In addition, we investigated the effects of PRAP1 reduction on the activation of checkpoint kinase 1 (Chk1) in response to thymidine. Chk1 mediates S-phase and G2 arrest in response to DNA damage.25, 26 Thymidine has previously been shown to induce S-phase arrest as a result of replication checkpoint activation through phospho-activation of Chk1.24 PRAP1 siRNA knockdown did not affect the activation (phosphorylation) of Chk1, indicating that the influence of PRAP1 on thymidine-induced S-phase arrest is unlikely to be mediated via replication checkpoint activation (Supplementary Figure S1C).
PRAP1 protects cells from caspase-dependent apoptosis induced by DNA-damaging agents
The abrogation of 5-FU-induced S-phase arrest by PRAP1 knockdown resulted in increased cell death. This sensitization of cancer cells to 5-FU by the loss of PRAP1 was effective even at a concentration of 1 μM 5-FU (Figure 5a). As p53 is known to induce apoptosis via a caspase-dependent pathway,27 we investigated whether the increased cell death observed is caspase-dependent. We found a significant increase in caspase 3 activity in PRAP1 knockdown cells treated with 10 μM 5-FU as compared with that of control siRNA cells (Figure 5b). Consistently, in PRAP1 knockdown cells, pre-treatment with either a pan-caspase inhibitor, Z-VAD, or a caspase 3-specific inhibitor blocked the enhanced cell death induced by 5-FU to the same level as that seen in untreated cells (Figure 5c). This demonstrates that PRAP1-knockdown cells undergo cell death by caspase-dependent apoptosis when exposed to 5-FU. The increased sensitivity of PRAP1 knockdown cells to 5-FU indicates that, in the absence of PRAP1, cells become more susceptible to DNA insults and apoptosis. Together, our results suggest that PRAP1 is able to modulate the p53-mediated response of cells to the cytotoxicity of genotoxic agents and may protect cells from apoptosis by inducing cell-cycle arrest.

PRAP1 knockdown sensitizes cells to caspase-dependent apoptosis induced by 5-FU. HCT116 cells were transfected with either control siRNA or PRAP1 siRNAs for 48 h and followed by the addition of 5-FU at the indicated doses for 24 h. (a) The percentage of dead cells, indicated by sub-G1 proportion was determined by flow cytometry detection in FL2 channel after staining the cells with propidium iodide. Knockdown of PRAP1 expression enhances the cell death induced by 5-FU as compared with control siRNA-treated cells (*P<0.05). (b) PRAP1 knockdown increases caspase 3 activity induced by 10 μM 5-FU as compared with control siRNA-treated cells (*P<0.05). (c) PRAP1 knockdown enhances caspase-dependent apoptosis induced by 5-FU. The pan caspase inhibitor Z-VAD-FMK or Caspase 3-specific inhibitor was added 1 h before 5-FU treatment in PRAP1 knockdown cells. Inhibition of caspases significantly reduced the percentage of sub-G1 cells in PRAP1 knockdown cells, indicating that these cells die by caspase-dependent apoptosis (*P<0.05)
Discussion
We have previously shown that the gene expression of PRAP1 was regulated by epigenetic mechanisms, such as methylation and histone acetylation.10 Here, we demonstrate that in response to cellular stresses such as DNA damage, PRAP1 expression is significantly induced by p53, a key player mediating cellular responses to DNA damage. We identified two p53-response elements within intron 1 of the PRAP1 gene and show that these elements are responsive to p53 transcriptional activity during DNA damage conditions.
One of the consequences of increased p53 activity in response to genotoxic stress is the induction of cell-cycle arrest. The mechanisms of p53-induced cell-cycle arrest in DNA-damaged cells have largely been attributed to the activation of its downstream target gene, p21WAF1/CIP1.28, 29 p21WAF1/CIP1 protein is a CDK inhibitor that inhibits CDK2 and CDK1 and their respective cyclin partners E and B, which are required for the progression of cell cycle from G1-S and from G2-M phases, respectively. Our data suggests that p53 may induce cell-cycle arrest in S-phase through PRAP1 induction and that loss of PRAP1 promotes apoptosis in 5-FU-treated cells. p53 can activate genes such as GADD45 to initiate the repair of damaged DNA or activate pro-apoptotic genes such as Bax to remove the damaged cells, which are beyond repair.30 p53-mediated activation of cell-cycle arrest may provide survival advantages; increased apoptosis and/or sensitivity to radiation were observed in tumors arising from p21-null mice.31, 32 Besides the p21 response, p53-activated PRAP1 may provide an alternative survival advantage through S-phase arrest, and this may be especially relevant in cells with defective signaling pathways, leading to the loss of ability to undergo G1 or G2 arrest.33 The ability of p53 to mediate multiple checkpoints in different phases of the cell cycle is crucial for its role in maintaining genomic stability. Thus far, the mechanisms by which PRAP1 contributes to cell-cycle arrest in the presence of DNA damage are unknown. There are no conserved domains identified for PRAP1 using various search tools. Our unpublished data reveal that PRAP1 binds to heat shock protein 70 (Hsp70), which has been shown to regulate the activities of key proteins involved in cell-cycle machinery such as p53 and CDK-434 and inhibit apoptosis.35 Interestingly the expression of Hsp70 was reported to be increased by 5-FU treatment in colon cancer cells,36 as well as other cellular stresses, including heat shock and ultraviolet exposure.37, 38 The interactions between PRAP1 and Hsp70 can therefore be further explored as potential avenues by which PRAP1 influences the cell-cycle progression.
The pro-survival effects of wild-type p53 in cancer cells can counteract the cytotoxic effects of tumor therapies targeting DNA damage pathways,39, 40 raising much concern. Studies have reported that expression of p53-dependent proteins, which blocks the cell cycle, such as p21WAF1/CIP1 and macrophage inhibitory cytokine-1, protects cells against p53-mediated apoptosis,41, 42 consistent with our findings that PRAP1 promotes cell survival. Following exposure to genotoxic stresses such as irradiation and cytotoxic drugs, PRAP1 is induced in a dose-dependent manner. However, p53 has also been reported to be important in sensitizing a range of human cancer cells to DNA-damaging agents.8 Here, we showed that by diminishing the protective arm of p53, such as by knocking down the p53-dependent expression of PRAP1, the efficacy of chemotherapeutic drugs can be improved. The loss of PRAP1 leads to enhanced DNA damage, attenuation of S-phase arrest and enhanced caspase-dependent apoptosis in tumor cells treated with 5-FU. Hence, we propose that in tumor cells, which respond to chemotherapeutic drugs by augmenting p53-dependent survival pathways, the inhibition of p53-dependent survival genes, such as those involved in cell-cycle arrest, can be developed as a strategy to improve tumor cell killing.
Materials and Methods
Cell culture and materials
HCT116 and HepG2 were purchased from the American Type Culture Collection (Rockville, MD, USA). p53−/− derived HCT116 cell line was kindly provided by Dr Bert Vogelstein (Howard Hughes Medical Institute, Chevy Chase, MD, USA). HCT116 and p53−/− cell lines were cultured in modified McCoy 5A medium supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA). HepG2 cells were cultured in HEPES modified DMEM medium (Sigma, St Louis, MO, USA) supplemented with 10% fetal bovine serum. All cell lines were maintained in a humidified 37 °C incubator with 5% CO2.
For drug treatment, 0.3 × 106 cells were seeded in a 6-well plate and treated with 5-FU and CPT (Sigma). In all, 0.3 × 106 of p53−/− cells were seeded and transfected with p53 wild-type or mutant expression vector using Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA), then treated with 5-FU and CPT 24 h after transfection. Treatment with caspase inhibitors were performed for 1 h with the following reagents: pan-caspase inhibitor, Z-VAD-FMK (40 μM) or caspase-3 inhibitor, Z-DEVD-FMK (20 μM; BD Biosciences, San Diego, CA, USA) before incubating with 5-FU. To track the cell-cycle progression with 5-FU treatment using dual BrdU/7AAD staining (FITC BrdU Flow Kit, BD), cells were synchronized by culturing for 18 h in serum-free medium before addition of 10 μM of 5-FU in complete media for 24 h. Cells were then pulsed with BrdU for 30 min, fixed and stained with anti-BrdU antibody and 7-AAD according to the manufacturer’s instructions.
Cloning of PRAP1 p53-response elements
The two p53 binding elements at intron 1 of PRAP1 gene was cloned into pGL3-Promoter vector. The primers used were: forward, 5′-AGTGATTCTCCTGCCTCAGC; reverse, 5′-CACCCATAATCCCAGCACTT. The DNA template used was BAC clone (RP11-122K13). The amplified insert was purified using QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). The purified insert and pGL3-Promoter vector were subjected to restriction enzyme digestion and ligated and transformed into competent cells. Positive clones were picked and grown. The constructed plasmid was verified by restriction digestion and sequencing. The correctly constructed plasmid was grown to large culture and purified using HiSpeed Plasmid Midi Kit (Qiagen) for subsequent transfection usage.
Dual-luciferase reporter assay
Cells were seeded in a 6-well plate and transfected 24 h before transfection. Plasmids were transfected using Lipofectamine 2000 Transfection Reagent (Invitrogen), according to the manufacturer’s recommendations. In all, 0.5 μg pGL3-PRAP1 or pGL3-p21 (positive control) or pGL3-Promoter vector (negative control) were co-transfected with empty pCMV (Vector), pCMV-p53 (p53 WT) or pCMV-p53mt153 (p53 Mut) into p53−/− cells. pRL-TK plasmid containing a renilla luciferase expression component was used as an internal control and co-transfected at a ratio of 1 : 50. Cells were harvested 24 h after transfection, and luciferase activities were measured using Dual-Luciferase Reporter Assay kit (Promega, Madison, WI, USA) according to the manufacturer’s instruction. Briefly, cells were lyzed in passive lysis buffer for 15 min with shaking at room temperature. In all, 100 μl of LAR II was added to 20 μl of lysate and measured for firefly luciferase signal using a fluorometer. Next, 100 μl of Stop and Glo reagent was added and measured for renilla luciferase signal. The firefly luciferase signal was normalized to that of renilla luciferase signal for each individual well.
Western blot analysis
Total protein extraction was carried out using lysis buffer (6 M urea, 1% 2-mercaptoethanol, 50 mM Tris buffer at pH 7.4, 1% sodium dodecyl sulfate (SDS) in phosphate-buffered saline (PBS) at pH 7.4). For detection of PRAP1, GolgiPlug (BD Biosciences) was added 6 h before each harvest. Cytoplasmic proteins were extracted using a gentle lysis buffer (0.1% NP-40, 0.1% Triton X-100 in PBS at pH 7). Total proteins and cytoplasmic proteins were subjected to SDS-PAGE and transferred to a nitrocellulose membrane (Amersham Hybond C-Extra, GE Healthcare Bioscience, Buckinghamshire, UK). The membrane was then blocked with 5% (w/v) non-fat milk in PBS containing 0.1% Tween-20 (PBST). Next, the membrane was incubated with anti-cyclin E, anti-cyclin D and anti-p53 (Santa Cruz Biotechnologies Inc., Santa Cruz, CA, USA) and anti-PRAP1 antibodies for 1 h, followed by incubation with horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies for 1 h. The immunoreactions were detected by chemiluminescence (Super-Signal West Dura chemiluminescence system, Thermo Scientific, IL, USA).
Flow cytometry
Cell-cycle analysis was performed using flow cytometry. Briefly, cells were trypsinized and resuspended in PBS, followed by fixation in ice-cold 70% ethanol. Cellular DNA was stained with propidium iodide solution (PBS containing 50 mg/ml propidium iodide, 0.1% Triton X-100 and 100 mg/ml RNaseA) and analyzed using FACS Vantage SE Flow Cytometry system (Becton Dickinson, NJ, USA). The cell-cycle phase distribution was analyzed using WinMDI software (Scripps Research Institute, La Jolla, CA, USA). Alternatively, the FITC BrdU Flow Kit (BD) was used for BrdU/7AAD double staining and data were analyzed using FlowJo 7.6.5 (Tree Star Inc., Ashland, OH, USA).
BrdU labeling assay
Cells were transfected with PRAP1 siRNA or control siRNA and treated with 5-FU for 24 h. Cells were then labeled with 10 mM BrdU for 30min. Cells were fixed and permeabilized for BrdU detection with monoclonal BrdU antibody according to the manufacturer’s recommendations (BD Biosciences).
Gene silencing by RNA interference
ON-TARGETplus SMARTpool, Human TP53, was used to knockdown p53 (Thermo Scientific). Synthetic Stealth (PRAP1 and its control siRNAs) were purchased from Invitrogen. To generate PRAP1 siRNA sequence-specific control, a four base mutation (underlined) was made in the PRAP1 siRNAs. The sequences for the PRAP1 siRNA duplex are as follows: PRAP1 siRNA1, 5′ CCGGTTGTGGGTGATGCCAAA; control siRNA1, 5′ CCGATTATGGGTAATGCCGAA; PRAP1 siRNA2, AAGGTCCCTATCAAGATGCAA; control siRNA2, AAGGTACCGATAAAAATGCAA. In all, 0.15 × 106 cells were seeded in a 6-well plate 24 h before transfection. Transfection was carried out using Lipofectamine RNAiMax Reagent (Invitrogen). Briefly, 0.08 to 1 μl of the siRNA duplex (20 μM) and 3 μl of the Lipofectamine RNAiMax Reagent (Invitrogen) were each diluted in 250 μl of Opti-MEM I Reduced Serum Medium, incubated together for 10 min at room temperature and then added to each well containing cells in 500 μl of Opti-MEM I Reduced Serum Medium. The medium was replaced with regular culture medium after 4 h.
RNA extraction and reverse transcription PCR
Total RNA was isolated using the Qiagen RNeasy kit as described by the manufacturer. First-strand cDNA was synthesized using ImProm-II Reverse Transcriptase (Promega) and PCR reactions were performed at the following cycling parameters: 94 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s, with 40 cycles for PRAP1 detection and 20 cycles for GAPDH. Primers used were as follows: PRAP1 forward, 5′-ACATGAGGAGGCTCCTCCTGG and reverse, 5′-TGGCCTGCTGGGCTCTATTTGTCT; GAPDH forward, 5′-GATGACCTTGCCCACAGCCT and reverse, 5′-ATCTCTGCCCCCTCTGCTGA.
Quantitative real-time RT-PCR
The induction of PRAP1 and p21 mRNA by gamma irradiation was investigated using real-time RT-PCR. RNA extraction and cDNA synthesis were as described above. PCR analysis of the cDNA was carried out using LightCycler-RNA Amplification kit SYBR Green I (Roche, Indianapolis, IN, USA) and real-time RT-PCR was performed in the LightCycler 480 Probe Master (Roche) according to the manufacturer’s recommendations. The following primers were used: PRAP1 forward (5′-CAGAGCAGGACCCAGAGAAG-3′), PRAP1 reverse (5′-AGAGTTTCGGCTTCTGGACA-3′), p21 forward (5′-GACACCACTGGAGGGTGACT-3′), p21 reverse (5′-CAGGTCCACATGGTCTTCCT-3′); GAPDH forward (5′-GAAGGTGAAGGTCGGAGTC-3′); GAPDH reverse (5′-GAAGATGGTGATGGGATTTC-3′). Amplification was achieved using the thermocycling parameters as follows: 95 °C for 30 s for 1 cycle; 95 °C for 30 s, 60 °C for 15 s and 72 °C for 15 s for 40 cycles. cDNA levels during the linear phase of amplification were normalized against GAPDH controls.
Colony-formation assay
Cells were transfected with siRNA as described above and treated with 5-FU for 24 h before re-seeding into 6-well plates. Cells were then cultured for 7 days and stained with 0.5% crystal violet in 30% ethanol and 3% formaldehyde for 10 min at room temperature, followed by four washings with water. Colonies were imaged and counted using the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Caspase 3 activity assay
Cells were transfected with PRAP1 siRNA or control siRNA for 48 h before treatment with 5-FU for 24 h. The cells were lyzed in 80 μl of ice-cold lysis buffer containing 50 mM HEPES, 100 mM NaCl, 0.1% CHAPS, 1 mM DTT and 0.1 mM EDTA, pH 7.4 for 5 min. Cells were then pelleted by centrifugation at 10 000 × g for 15 min at 4 °C. In all, 20 μl of the supernatant was added to 100 μl assay buffer containing 50 mM HEPES, 100 mM NaCl, 0.1% CHAPS, 1 mM DTT and 0.1 mM EDTA, 10% glycerol, pH 7.4 and 10 μl of caspase substrate was then added to the reaction system to achieve a final substrate concentration of 0.2 mM. The cleavage of substrate was measured using spectrophotometric detection at 405 nm, and the activities were calculated according to the manufacturer’s instructions (Roche).
Micronucleus assay
Micronucleus assay measures the chromosomal breakage that occurs due to exposure to genotoxic agents. After treatment with 5-FU, cells were then trypsinized and subsequently fixed using Camoy’s fixative (acetic acid/methanol, 1 : 3) with 3.7% formaldehyde. Fixed cells were washed three times with Camoy’s fixative. Cells were dropped onto clean slides and stained with acridine orange to differentially stain the cytoplasm and nucleus. One thousand mono-nucleated cells were scored for each sample.
Alkaline single-cell gel electophoresis (comet) assay
DNA damage was determined by the comet assay.43 Briefly, cells were treated with 5-FU and harvested by trypsinization, then resuspended in Hanks’ Balanced Salt Solution with 10% DMSO with EDTA. Cells were then added to 0.75% (w/v) low melting point agarose and transferred onto comet slides (Trevigen, Gaithersburg, MD, USA). Slides were incubated in hydrolysis buffer (2.5 M NaCl, 0.1 M EDTA, 10 mM Tris Base, pH 10) with 1% Triton X for 1 h. Cells were then subjected to electrophoresis, followed by neutralization in 0.5 M Tris-HCl, pH 7.5, drying and staining with SYBR green dye. One hundred comets were analyzed per sample. The extent of DNA damage was expressed as tail movement, which corresponds to the fraction of DNA in the tail of the comet. The tail movement was captured and analyzed using the MetaSystems CometImager software version 1.2 (MetaSystems, Altussheim, Germany).
ChIP assay
HCT116 cells were treated or untreated with 5-FU at 25 μM or 50 μM for 24 h. Cells were fixed and chromatin was sheared by sonication and immunoprecipitated overnight with/without 1 μl of anti-p53 antibody of ChIP grade (Millipore, Billerica, MA, USA) with a ChIP assay kit (Millipore). The co-immunoprecipitated promoter fragments and input DNA were amplified by PCR. The primers used for the first p53-response element in intron 1 of PRAP1 gene are: ChIP P1-forward: 5′ ACCACGCCTGGCTAATTTTT and -reverse: 5′ GAGGCAACAGATCACTTGAGG; and for the second p53-response elements are: ChIP P2-forward: 5′ CTGGGATTACAGGCGTGAGT and -reverse: 5′ TCACCTGAGGTCAGGAGTTTG. Another three pairs of primers, which recognize more than 600 bp upstream and downstream of the two p53-response elements, were used to amplify the PRAP1 promoter as negative controls. The primers were: N1-forward: 5′ CTCTTCCCCCTCCTCTAAG and -reverse: 5′ CTCTTCCCCCTCCTCTAAG; N2-forward: 5′ ACCCATGTGCAAGTGTGTTC and -reverse: 5′ GTTGGCTTGGAAACTCCAGA; N3-forward: 5′ TGTCCGAACTGACCTGACAC and -reverse: 5′ CACCTGCTGTGTCCCAGA. The primers used to amplify the known p53-response element in the p21 promoter as a positive control were: p21-forward: 5′ GTGGCTCTGATTGGCTTTCTG; and -reverse: 5′ CTGAAAACAGGCAGCCCAAG.
Statistical analysis
Non-paired Student’s t-test was used to compare the means of two groups and statistical significance was set at P<0.05.
Abbreviations
- PRAP1:
-
proline-rich acidic protein 1
- 5-FU:
-
5-fluorouracil
- CPT:
-
camptothecin
- CDK:
-
cyclin-dependent kinase
- Chk1:
-
checkpoint kinase 1
- BrdU:
-
5-bromo-2′deoxyuridine
- 7-AAD:
-
7-amino-actinomycin D
- PBS:
-
phosphate-buffered saline
- ChIP:
-
chromatin immunoprecipitation
- siRNA:
-
small-interfering RNA
References
Hakem R . DNA-damage repair; the good, the bad, and the ugly. EMBO J 2008; 27: 589–605.
Piette J, Neel H, Marechal V . Mdm2: keeping p53 under control. Oncogene 1997; 15: 1001–1010.
Harris SL, Levine AJ . The p53 pathway: positive and negative feedback loops. Oncogene 2005; 24: 2899–2908.
Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992; 71: 587–597.
Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T . p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993; 362: 847–849.
Lane DP . Exploiting the p53 pathway for cancer diagnosis and therapy. Br J Cancer 1999; 80 (Suppl 1): 1–5.
Pestell KE, Hobbs SM, Titley JC, Kelland LR, Walton MI . Effect of p53 status on sensitivity to platinum complexes in a human ovarian cancer cell line. Mol Pharmacol 2000; 57: 503–511.
Park CM, Park MJ, Kwak HJ, Moon SI, Yoo DH, Lee HC et al. Induction of p53-mediated apoptosis and recovery of chemosensitivity through p53 transduction in human glioblastoma cells by cisplatin. Int J Oncol 2006; 28: 119–125.
Kasik J, Rice E . A novel complementary deoxyribonucleic acid is abundantly and specifically expressed in the uterus during pregnancy. Am J Obstet Gynecol 1997; 176: 452–456.
Zhang J, Wong H, Ramanan S, Cheong D, Leong A, Hooi SC . The proline-rich acidic protein is epigenetically regulated and inhibits growth of cancer cell lines. Cancer Res 2003; 63: 6658–6665.
De Angelis PM, Svendsrud DH, Kravik KL, Stokke T . Cellular response to 5-fluorouracil (5-FU) in 5-FU-resistant colon cancer cell lines during treatment and recovery. Mol Cancer 2006; 5: 20.
Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y . Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J 1999; 18: 1397–1406.
Gonzalez C, Najera O, Cortes E, Toledo G, Lopez L, Betancourt M et al. Hydrogen peroxide-induced DNA damage and DNA repair in lymphocytes from malnourished children. Environ Mol Mutagen 2002; 39: 33–42.
Yau T, Chan P, Ching Chan Y, Wong BC, Liang R, Epstein RJ . Review article: current management of metastatic colorectal cancer—the evolving impact of targeted drug therapies. Aliment Pharmacol Ther 2008; 27: 997–1005.
Morgan WF, Day JP, Kaplan MI, McGhee EM, Limoli CL . Genomic instability induced by ionizing radiation. Radiat Res 1996; 146: 247–258.
Saramaki A, Banwell CM, Campbell MJ, Carlberg C . Regulation of the human p21(waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res 2006; 34: 543–554.
Hoh J, Jin S, Parrado T, Edington J, Levine AJ, Ott J . The p53MH algorithm and its application in detecting p53-responsive genes. Proc Natl Acad Sci USA 2002; 99: 8467–8472.
Fenech M, Holland N, Chang WP, Zeiger E, Bonassi S . The HUman MicroNucleus Project—An international collaborative study on the use of the micronucleus technique for measuring DNA damage in humans. Mutat Res 1999; 428: 271–283.
Riley T, Sontag E, Chen P, Levine A . Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9: 402–412.
Maybaum J, Ullman B, Mandel HG, Day JL, Sadee W . Regulation of RNA- and DNA-directed actions of 5-fluoropyrimidines in mouse T-lymphoma (S-49) cells. Cancer Res 1980; 40: 4209–4215.
Takeda H, Haisa M, Naomoto Y, Kawashima R, Satomoto K, Yamatuji T et al. Effect of 5-fluorouracil on cell cycle regulatory proteins in human colon cancer cell line. Jpn J Cancer Res 1999; 90: 677–684.
Phoolcharoen W, Smith DR . Internalization of the dengue virus is cell cycle modulated in HepG2, but not Vero cells. J Med Virol 2004; 74: 434–441.
St-Denis NA, Derksen DR, Litchfield DW . Evidence for regulation of mitotic progression through temporal phosphorylation and dephosphorylation of CK2alpha. Mol Cell Biol 2009; 29: 2068–2081.
Rodriguez R, Meuth M . Chk1 and p21 cooperate to prevent apoptosis during DNA replication fork stress. Mol Biol Cell 2006; 17: 402–412.
Zhao H, Watkins JL, Piwnica-Worms H . Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci USA 2002; 99: 14795–14800.
Sorensen CS, Syljuasen RG, Falck J, Schroeder T, Ronnstrand L, Khanna KK et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003; 3: 247–258.
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303: 1010–1014.
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282: 1497–1501.
Waldman T, Kinzler KW, Vogelstein B . p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 1995; 55: 5187–5190.
Velculescu VE, El-Deiry WS . Biological and clinical importance of the p53 tumor suppressor gene. Clin Chem 1996; 42 (6 Pt 1): 858–868.
Wang YA, Elson A, Leder P . Loss of p21 increases sensitivity to ionizing radiation and delays the onset of lymphoma in atm-deficient mice. Proc Natl Acad Sci USA 1997; 94: 14590–14595.
Bearss DJ, Lee RJ, Troyer DA, Pestell RG, Windle JJ . Differential effects of p21(WAF1/CIP1) deficiency on MMTV-ras and MMTV-myc mammary tumor properties. Cancer Res 2002; 62: 2077–2084.
Al-Khalaf HH, Lach B, Allam A, AlKhani A, Alrokayan SA, Aboussekhra A . The p53/p21 DNA damage-signaling pathway is defective in most meningioma cells. J Neurooncol 2007; 83: 9–15.
Zylicz M, King FW, Wawrzynow A . Hsp70 interactions with the p53 tumour suppressor protein. EMBO J [Research Support, Non-U.S. Gov’t Review] 2001; 20: 4634–4638.
Li CY, Lee JS, Ko YG, Kim JI, Seo JS . Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and upstream of caspase-3 activation. J Biol Chem [Research Support, Non-U.S. Gov’t] 2000; 275: 25665–25671.
Grivicich I, Regner A, Zanoni C, Correa LP, Jotz GP, Henriques JA et al. Hsp70 response to 5-fluorouracil treatment in human colon cancer cell lines. Int J Colorectal Dis 2007; 22: 1201–1208.
Jonak C, Klosner G, Trautinger F . Heat shock proteins in the skin. Int J Cosmet Sci 2006; 28: 233–241.
Matsuda M, Hoshino T, Yamashita Y, Tanaka K, Maji D, Sato K et al. Prevention of UVB radiation-induced epidermal damage by expression of heat shock protein 70. J Biol Chem [Research Support, Non-U.S. Gov’t] 2010; 285: 5848–5858.
McKay BC, Becerril C, Ljungman M . P53 plays a protective role against UV- and cisplatin-induced apoptosis in transcription-coupled repair proficient fibroblasts. Oncogene 2001; 20: 6805–6808.
Smith ML, Fornace AJ . p53-mediated protective responses to UV irradiation. Proc Natl Acad Sci USA 1997; 94: 12255–12257.
Staversky RJ, Vitiello PF, Gehen SC, Helt CE, Rahman A, Keng PC et al. p21(Cip1/Waf1/Sdi1) protects against hyperoxia by maintaining expression of Bcl-X(L). Free Radic Biol Med 2006; 41: 601–609.
Agarwal MK, Hastak K, Jackson MW, Breit SN, Stark GR, Agarwal ML . Macrophage inhibitory cytokine 1 mediates a p53-dependent protective arrest in S phase in response to starvation for DNA precursors. Proc Natl Acad Sci USA 2006; 103: 16278–16283.
Oliveira RJ, Matuo R, da Silva AF, Matiazi HJ, Mantovani MS, Ribeiro LR . Protective effect of beta-glucan extracted from Saccharomyces cerevisiae, against DNA damage and cytotoxicity in wild-type (k1) and repair-deficient (xrs5) CHO cells. Toxicol In Vitro 2007; 21: 41–52.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by P Salomoni
Supplementary Information accompanies the paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Huang, B., Zhuo, J., Leung, C. et al. PRAP1 is a novel executor of p53-dependent mechanisms in cell survival after DNA damage. Cell Death Dis 3, e442 (2012). https://doi.org/10.1038/cddis.2012.180
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.180
Keywords
This article is cited by
-
Molecular intervention of colon cancer and inflammation manifestation by tannin capped biocompatible controlled sized gold nanoparticles from Terminalia bellirica: A green strategy for pharmacological drug formulation based on nanotechnology principles
3 Biotech (2021)
-
The pro-survival function of DLEC1 and its protection of cancer cells against 5-FU-induced apoptosis through up-regulation of BCL-XL
Cytotechnology (2019)
-
p73 regulates serine biosynthesis in cancer
Oncogene (2014)
-
miR-16 and miR-26a target checkpoint kinases Wee1 and Chk1 in response to p53 activation by genotoxic stress
Cell Death & Disease (2013)
-
Tumor suppressor WWOX binds to ΔNp63α and sensitizes cancer cells to chemotherapy
Cell Death & Disease (2013)

{kind=link}