
Abstract
The mammalian target of rapamycin (mTOR) is a key regulator of cell growth, autophagy, translation, and survival. Dysregulation of mTOR signaling is associated with cancer, diabetes, and autism. However, a role for mTOR signaling in neuronal death is not well delineated. Here we show that global ischemia triggers a transient increase in mTOR phosphorylation at S2448, whereas decreasing p-mTOR and functional activity in selectively vulnerable hippocampal CA1 neurons. The decrease in mTOR coincides with an increase in biochemical markers of autophagy, pS317-ULK-1, pS14-Beclin-1, and LC3-II, a decrease in the cargo adaptor p62, and an increase in autophagic flux, a functional readout of autophagy. This is significant in that autophagy, a catabolic process downstream of mTORC1, promotes the formation of autophagosomes that capture and target cytoplasmic components to lysosomes. Inhibitors of the lysosomal (but not proteasomal) pathway rescued the ischemia-induced decrease in mTOR, consistent with degradation of mTOR via the autophagy/lysosomal pathway. Administration of the mTORC1 inhibitor rapamycin or acute knockdown of mTOR promotes autophagy and attenuates ischemia-induced neuronal death, indicating an inverse causal relation between mTOR, autophagy, and neuronal death. Our findings identify a novel and previously unappreciated mechanism by which mTOR self-regulates its own levels in hippocampal neurons in a clinically relevant model of ischemic stroke.
Similar content being viewed by others


Main
Mammalian target of rapamycin (mTOR) is a serine/threonine kinase that integrates external cues and is a central regulator of cell growth, proliferation, protein synthesis, autophagy and survival.1, 2, 3 mTOR forms two macromolecular complexes, mTOR complex1 (mTORC1) and 2, which have distinct functions and downstream targets.4 Whereas mTORC1 is a central regulator of cap-dependent translation,5 mTORC2 is a pivotal regulator of the actin cytoskeleton, spine structure and memory.6 In neurons, mTORC1 is strategically positioned at pre- and postsynaptic sites where it influences synaptic plasticity by regulating the balance between protein synthesis and degradation via the autophagy/lysosomal7, 8, 9 and ubiquitin/proteasomal pathways.10 Under nutrient-rich conditions, mTORC1 serves as a brake on autophagy,11, 12, 13, 14 a process of programmed catabolic activity and cell survival, which is increased during periods of cellular stress.14 In response to stress, mTOR translocates to the surface of lysosomes, where Rheb, a small GTPase, strategically positions mTOR in close proximity to Akt and p70S6 kinase, which phosphorylate mTOR at S2448.15, 16, 17, 18
Activation of mTOR is essential for the brake imposed on autophagy by inhibition of downstream targets critical to initiation of autophagy.12 Upon activation, mTOR phosphorylates and inactivates Unc-51-like autophagy-activating kinase 1 (ULK-1) at S757, preventing assembly of ULK-1 with scaffolding protein Atg13 and adhesion protein FIP200.11 This, in turn, prevents phosphorylation and activation of Beclin-1 at S14 and halts autophagy.13 Beclin-1 is a signaling protein downstream of mTORC1 and critical to the ‘nucleation phase’ of autophagy.12, 19 Beclin-1 promotes lipidation of LC3-I to generate its lipidated form LC3-II and formation of autophagosomes. Upon lipidation, LC3-II localizes to the autophagosome membrane, where it promotes formation of autophagosomes. Autophagosomes deliver proteins and organelles to lysosomes for degradation.
In response to cellular stress, phosphorylation of mTOR at S2448 is reduced and the brake imposed on autophagy is released. Phosphorylation of ULK-1 at S757 is decreased, enabling ULK-1 to be released by mTOR and to assemble with AMPK, which phosphorylates and activates ULK-1 at S317. This, in turn, activates Beclin-1 and promotes lipidation of LC3-I to generate LC3-II. LC3-II localizes to the autophagosome membrane, enabling elongation of the limiting membrane, which seals to form LC3-II-(+) autophagosomes. Autophagosomes degrade cargo adaptors such as p62 via autophagy. Growing evidence indicates that dysregulation of mTOR is associated with human diseases, including cancer, diabetes, and autism.1, 2, 20, 21, 22, 23 However, a role for mTOR in ischemia-induced neuronal death is, as yet, unclear.
Transient global ischemia elicits selective, delayed death of primarily CA1 pyramidal neurons and impaired cognition, evident at 5–7 days after insult.24, 25, 26, 27 At the cellular level, ischemia promotes translational arrest and accumulation of polyubiquitinated proteins,28 indicating a shift in the balance away from protein synthesis and toward protein degradation. In the first 1–3 h following induction of ischemia, the functional integrity of the mitochondrial membrane is compromised, leading to the release of cytochrome c and caspase activation.29 This is significant in that caspases cleave and inactivate Beclin-1, thereby destroying its proautophagic activity.
The present study was undertaken to examine a role for mTOR signaling and autophagy in ischemia-induced neuronal death. Here we show that mTOR is decreased in CA1 (but not CA3) as early as 3 h after ischemia. The decrease in mTOR precedes or coincides with an increase in markers of autophagy, pS317-ULK-1, pS14-Beclin-1, and LC3-II/I ratio (indicator of autophagic flux), and a decrease in the ubiquitin-binding cargo adaptor p62, indicating induction of autophagy in neurons destined to die. RNAi-mediated depletion of mTOR or inhibition of mTORC1 by rapamycin before ischemia activates autophagy, which promotes degradation of mTOR and attenuates ischemia-induced neuronal death. These findings demonstrate a causal relation between loss of mTOR, activation of autophagy, and neuronal death, and reveal a novel mechanism whereby mTOR self-regulates its own degradation via the autophagy/lysosomal pathway in a clinically relevant model of ischemia.
Results
Global ischemia suppresses mTOR abundance and activity in the CA1
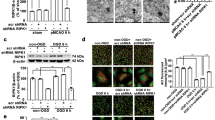
We first examined the impact of global ischemia on mTOR abundance and phosphorylation in the selectively vulnerable CA1. Toward this end, we subjected adult male rats to global ischemia or sham operation and assessed mTOR abundance and phosphorylation at S2448, a target of Akt and p70S6 kinase, at times after ischemia. We focused on S2448 because phosphorylation at this site is implicated in mTORC1 activity and suppression of autophagy.12, 30 Ischemia elicited a transient increase in phosphorylation of mTOR at S2448 and a decrease in mTOR abundance in CA1, each evident by 3 h (Figure 1a). In the resistant CA3, p-mTOR and mTOR were unchanged (Supplementary Figure 1a).

Global ischemia transiently increases mTOR phosphorylation and decreases mTOR abundance and activity in CA1. (a) Top: Representative western blot showing that ischemia transiently increases phosphorylation of mTOR at S2448, as early as 3 h, whereas decreasing mTOR abundance, as early as 3 h and as late as 24 h, in the CA1. Accordingly, abundance of pS2448-mTOR goes down in insulted neurons by 24 h. Bottom: Summary data. (b) Immunolabeling of hippocampal cross-sections from animals subjected to global ischemia or sham operation demonstrate that mTOR abundance is decreased in CA1 (arrowheads) but not CA3 or DG, assessed at 24 h following ischemia. Importantly, mTOR abundance decreases in CA1 pyramidal neurons marked by NeuN. Left: × 2 image, scale bar=500 μm. Middle: × 20 image, right: x63 image, scale bar=5 μm. (c and d) Top: Representative western blot showing that ischemia decreases p-T37/46-4E-BP (c) and pS757-ULK-1 (d), functional readouts of mTOR kinase activity in CA1, as early as 3 h and as late as 24 h after ischemia. Bottom: Summary data; n=4–8 animals per treatment group/3 independent experiments. (e) Top: Representative western blot showing that ischemia increases p-T172-AMPK in CA1, evident at 3 h and 6 h after ischemia. Bottom: Summary data. (f) Top: Representative western blot showing that ischemia increases pS317-ULK-1, a functional readout of AMPK activity in CA1, as early as 3 h and as late as 24 h after ischemia. Here and in all figures, data represent mean±S.E.Ms. ***P<0.001, **P<0.01, and *P<0.05. (g) Model depicting a hypothetical mechanism by which ischemia downregulates mTOR abundance (and activity) in CA1 neurons, which, in turn, removes the brake on autophagy imposed by mTORC1 under physiological conditions, enabling phosphorylation of ULK-1 at S317 by AMPK to initiate autophagy
These findings indicate that the ischemia-induced increase in p-mTOR and reduction in total mTOR are subfield-specific, but do not distinguish between neuronal and non-neuronal cells. To address this, we performed immunohistochemistry on brain sections. mTOR was markedly decreased in CA1 pyramidal neurons marked by NeuN, but not in adjacent CA3 pyramidal or dentate gyrus granule cells, at 24 h after ischemia (Figure 1b). Ischemia did not detectably alter mTOR mRNA at any time examined (Supplementary Figure 1b), consistent with regulation mTOR at the post-transcriptional level.
We next addressed the impact of ischemia on two functional readouts of mTORC1 activity, phosphorylation of 4E binding protein (4E-BP), a direct downstream target of mTORC1 and negative regulator of cap-dependent translation,31 and ULK-1 (aka Atg1), a protein involved in the initiation of autophagy,12 at S757. Whereas phosphorylation of 4E-BP promotes initiation of translation, phosphorylation of ULK-1 at S757 sequesters ULK-1 by stabilizing its association with mTOR,32 and thereby halts autophagy. Ischemia induced a decrease in p-4E-BP at T37/46 (Figure 1c) and p-ULK-1 at S757 (Figure 1d).
The results thus far show that the ischemia-induced reduction in mTOR coincides with a reduction in phosphorylation of mTOR downstream targets 4E-BP and ULK-1, but do not address a causal relation between them. Toward this end, we applied rapamycin, which reduces mTOR activity, but not abundance. Rapamycin reduced p-mTOR (not illustrated) and mTOR targets ULK-1, 4E-BP, and S6 under physiological conditions and further reduced p-ULK-1 under ischemic conditions. Thus, inhibition of mTOR, even in the absence of a neuronal insult, is sufficient to induce autophagy (Supplementary Figure 2a). Next, we applied small interfering RNA (siRNA) to mTOR, which reduces mTOR abundance without affecting phosphorylation. siRNA-mediated knockdown of mTOR reduced mTOR abundance and diminished pS757-ULK-1 and p-T37/46-4E-BP as well as p-S 240/244-S6 (Supplementary Figure 2b). Thus, a decrease in mTOR, even in the absence of a neuronal insult, is sufficient to promote dephosphorylation of ULK-1 at S757, a key step in initiation of autophagy. The lack of effect of ischemia on pS6 is consistent with findings that pS6 is under the control not only of mTOR but also ERK/p90RSK.33
Adenosine monophosphate (AMP)-activated protein kinase (AMPK), a kinase activated by stressors,34 is also a trigger of autophagy. Dephosphorylation of ULK-1 at S757 releases ULK-1 from mTOR, enabling it to associate with AMPK. AMPK phosphorylates and activates ULK-1 at S317, and thereby promotes the initiation of autophagy.32, 35 Ischemia induced an increase in p-AMPK- and AMPK-dependent phosphorylation of ULK-1 at S317, evident at 3 h and as late as 24 h, the latest time examined (Figures 1e and f). These findings are consistent with a model whereby ischemia downregulates mTOR in CA1 neurons, and thereby removes the brake on ULK-1, enabling AMPK to phosphorylate ULK-1 and initiate autophagy. The decrease in mTOR abundance coincides with the increase in AMPK activity, suggesting that mTOR and AMPK act in concert to promote the onset of autophagy.
Global ischemia upregulates biochemical markers of autophagy in CA1
We next addressed the impact of ischemia on induction of autophagy. Ischemia induced phosphorylation and activation of Beclin-1 at S14, a protein critical to the nucleation phase of autophagy, in CA1, evident at 6 h after ischemia, with little or no change in Beclin-1 abundance (Figure 2a). We next examined the impact of ischemia on steady-state levels of autophagic markers. Ischemia markedly increased LC3-II and LC3-II/LC3-I, an indicator of the number of autophagosomes (Figure 2b). To assess the impact of ischemia on the release of cargo from autophagosomes to lysosomes, we measured the abundance of p62. Ischemia significantly decreased p62, which facilitates the transfer of cargo from the cytosol to autophagosomes (Figure 2c), first evident at 3 h after ischemia and as late as 24 h. These findings indicate an enhanced autophagic flux, a functional readout of autophagy. Thus, the ischemia-induced decrease in mTOR precedes or coincides with increased p-Beclin-1 and LC3-II/I and decreased p62.

Global ischemia promotes an increase in markers of autophagy in CA1. (a) Top: Representative western blot showing that global ischemia enhances phosphorylation of Beclin-1 at S14, evident at 6 h, with little or no change in Beclin-1 abundance. Bottom: Summary data. (b) Top: Representative western blot demonstrating that ischemia enhances LC3-II, as early as 3 h and as late as 24 h after insult. Bottom: Summary data. (c) Top: Representative western blot demonstrating that global ischemia decreases expression of the cargo adapter p62, as early as 3 h and as late as 24 h after insult. Bottom: Summary data; n=4–8 animals per treatment group/3 independent experiments. (d) Western blot analysis showing that mTOR protein translocates from the cytoplasmic fraction (cyto) to the lysosomal-associated membrane protein 2 (LAMP2)-enriched lysosomal fraction (lyso) isolated from lysates of CA1 at 24 h after global ischemia in vivo. ***P<0.001, **P<0.01, and *P<0.05
We next examined whether ischemia promotes redistribution of mTOR within CA1 neurons. Ischemia did not detectably alter mTOR abundance in the membrane-enriched fraction (Supplementary Figure 3a), but increased mTOR in intracellular vesicles marked by lysosomal-associated membrane protein 2, assessed by western blot (Figure 2d) and immunocytochemistry (Supplementary Figures 3b and c). Thus, ischemia promotes translocation of mTOR to the outer surface of the lysosome, where it is positioned in close proximity to upstream kinases.
OGD mimics global ischemia
The results thus far indicate that ischemia decreases mTOR abundance and increases markers of autophagy, but do not identify the molecular mechanisms underlying these changes. To address this, we took advantage of oxygen-glucose deprivation (OGD), a well-established in vitro model of global ischemia in hippocampal neurons.36, 37, 38 Cellular models afford more precise control over the physicochemical milieu than is possible in the intact animal. OGD decreased mTOR (Figure 3a) and increased markers of autophagy, resembling the molecular changes induced by ischemia in vivo. OGD markedly decreased pS757-ULK-1 (Figure 3b), increased p-Beclin-1 and LC3-II, and decreased p62 (Figures 3c–e). Moreover, OGD decreased the LC3-II/I ratio, a readout of autophagic flux (Figure 3f and g). Thus, OGD mimics the impact of global ischemia on mTOR and autophagy in CA1 in vivo. The decrease in mTOR abundance was specific to OGD, in that other paradigms known to induce neuronal death, such as staurosporine-induced apoptosis or hydrogen peroxide-induced oxidative stress, did not detectably alter mTOR (Supplementary Figure 4).

OGD, an in vitro model of global ischemia, decreases mTOR and increases markers of autophagy in hippocampal neurons. (a) Top: Representative western blot showing that OGD in cultured neurons decreases mTOR protein levels, evident at 3 h and as late as 24 h after insult. Bottom: Summary data. (b) OGD in cultured neurons decreases phosphorylation of ULK-1 at S757, a downstream target of mTOR. Bottom: Summary data. (c) Top: Representative western blot demonstrating that OGD enhances phosphorylation of Beclin-1 at S14, but has little or no effect on total Beclin-1 abundance. Bottom: Summary data. (d) Top: Representative western blot demonstrating that OGD enhances LC3-II abundance, as early as 3 h and as late as 24 h after insult. Bottom: Summary data. (e) Top: Representative western blot demonstrating that OGD reduces abundance of p62, evident at 3 h and as late as 24 h after insult. Bottom: Summary data; n=4–8 coverslips per treatment group/3 independent experiments. (f) Top: Representative western blot demonstrating that OGD enhances LC3-II abundance even greater in the presence of lysosomal inhibitors leupeptin and ammonium chloride. Bottom: Summary data; n=6 coverslips per treatment group/2 independent experiments. (g) Summary data illustrating the average abundance of LC3 in the presence of lysosomal inhibitors/average LC3 abundance in the presence of vehicle, a measure of autophagy flux in neurons. ***P<0.001, **P<0.01, and *P<0.05
Inhibition of the lysosome rescues the ischemia-induced decrease in mTOR
To determine the mechanism by which ischemia decreases mTOR, we induced OGD in hippocampal neurons in the presence or absence of inhibitors of lysosomal and proteasomal degradation and assessed mTOR abundance 24 h after insult. OGD induced a marked decrease in mTOR abundance, evident at 24 h. Whereas inhibition of lysosomal degradation by treatment with leupeptin/ammonium chloride did not detectably alter mTOR abundance under normoxic conditions, it rescued the ischemia-induced decrease in mTOR (Figure 4a). Inhibition of proteasomal degradation by lactacystin39 modestly decreased basal mTOR, but did not prevent the OGD-induced decrease in mTOR (Figure 4b). Thus, mTOR is preferentially degraded by the autophagy/lysosomal pathway in insulted hippocampal neurons. Moreover, treatment of neurons with rapamycin, an inhibitor of mTORC1 and known activator of the lysosomal pathway, had little or no effect on mTOR under normoxic or ischemic conditions (Figure 4c). These findings indicate that the ischemia-induced decrease in mTOR does not require mTOR activity.

The ischemia-induced decrease in mTOR is rescued by lysosomal (but not proteasomal) inhibitors. (a) Top: Representative western blot demonstrating that the lysosome inhibitors ammonium chloride (10 mM) and leupeptin (50 μM for 3 h) prevent the ischemia-induced decrease in mTOR, assessed at 24 h following insult. Bottom: Summary data. (b) Top: Representative western demonstrating that the proteasome inhibitor lactacystin (2.5 μM) enhances LC3-II abundance but does not prevent the ischemia-induced decrease in mTOR abundance at 24 h after insult. Bottom: Summary data. (c) Top: Representative western blot demonstrating that the mTOR kinase inhibitor rapamycin (50 nM) does not prevent the ischemia-induced decrease in mTOR abundance at 24 h after insult. Bottom: Summary data. (d) Top: Representative western showing that intracerebroventricular injection of the lysosomal protease inhibitor leupeptin (10 mM) 1 h before induction of global ischemia blocks lysosomal degradation, as assessed by an upregulation of p62 expression, and prevents the ischemia-induced decrease in mTOR protein abundance at 6 and 24 h following ischemic insult. Bottom: Summary data. (e) Top: Representative western blot showing that the autophagy inhibitor C43 attenuates OGD-induced degradation of mTOR, assessed at 24 h following insult. Bottom: Summary data. (f) Representative western blot showing that transfection of hippocampal neurons with siRNA targeting Atg7, but not NT siRNA, knocks down Atg7 protein to ~2% of control levels and attenuates OGD-induced degradation of mTOR at 24 h after insult (g). (h) Representative western blot showing that ischemia reduces Raptor abundance, but loss of Raptor is not blocked by C43. Bottom: Summary data; n=4–6 coverslips or animals per treatment group in three independent experiments. ***P<0.001, **P<0.01, ##P<0.01, and *,#P<0.05
To verify a role for lysosomal degradation in the ischemia-induced decrease in mTOR in vivo, we administered the lysosomal inhibitor leupeptin into rats intracerebroventricularly 1 h before induction of ischemia. Whereas leupeptin did not detectably alter mTOR or p62 under normoxic conditions, it rescued the ischemia-induced decrease in mTOR and p62 (Figure 4d). These findings confirm that mTOR is preferentially degraded by the lysosomal pathway in postischemic CA1.
We next addressed a role for autophagy in mTOR degradation. To address this issue, we first examined the impact of C43 on ischemia-induced mTOR degradation. Application of C43, a well-established inhibitor of autophagy, to hippocampal neurons during and after ischemia diminished mTOR degradation (Figure 4e). Moreover, shRNA to Atg7, an E1-like enzyme essential for autophagosome formation,40 markedly reduced Atg7 (Figure 4f) and prevented ischemia-induced degradation of mTOR (Figure 4g). Collectively, these findings strongly suggest that autophagy is critical to ischemia-induced degradation of mTOR. Although ischemia also reduced Raptor, a binding partner of mTOR and defining component of mTORC1, degradation of Raptor was not blocked by C43 (Figure 4h), indicating that it is not mediated by autophagy. Thus, autophagy does not mediate degradation of all components of mTORC1.
mTOR is causally related to autophagy in neurons
The results thus far indicate that the ischemia-induced decrease in mTOR and p-mTOR coincide with an upregulation of autophagy, but do not address a causal relation between loss of mTOR and autophagy. To address this issue, we first exposed hippocampal neurons to rapamycin or vehicle 1 day before ischemia and assessed markers of autophagy by western blot. Application of rapamycin (24 h) reduced phosphorylation of ULK-1 at S757, increased p-Beclin-1, increased LC3-II, and decreased p62 (Figures 5a–e). These findings indicate that a reduction of mTOR activity, even in the absence of a neuronal insult, is sufficient to induce autophagy.

Loss of mTOR, even in the absence of a neuronal insult, is causally related to autophagy in neurons. (a) Representative western blots demonstrating that the mTOR inhibitor rapamycin (50 nM) administered for 24 h in the absence of ischemia upregulates markers of autophagy, as evidenced by a decrease in pS757-ULK-1, an increase in pS14-Beclin-1 and LC3-II/I, and a decrease in the cargo adapter p62. (b–e) Summary of data in (a). (f) Representative western blot showing that transfection of hippocampal neurons with siRNA targeting mTOR, but not NT siRNA, knocks down mTOR protein to ~30% of control levels and upregulates markers of autophagy (g–j). Summary data; n=5–8 coverslips per treatment group in three independent experiments. ***P<0.001, **P<0.01, and *P<0.05
Next, we treated neurons with siRNA to mTOR or non-targeting (NT) siRNA for 4 days before ischemia and assessed markers of autophagy by western blot. siRNA to mTOR, but not NT siRNA, decreased mTOR by ~75% and activated autophagy, as assessed by an increase in p-Beclin-1, an increase in LC3-II abundance, and a decrease in p62 (Figures 5f–j). These findings indicate that pharmacological inhibition or siRNA-mediated acute knockdown of mTOR, even in the absence of neurological insult, can activate markers of autophagy.
Acute inhibition or RNAi-mediated depletion of mTOR attenuates neuronal death
The results thus far indicate that the ischemia-induced decrease in mTOR and p-mTOR are causally related to activation of autophagy, but do not address a causal relation between ischemia-induced loss of mTOR and neuronal death. Toward this end, we undertook two experimental strategies. First, we exposed hippocampal neurons to rapamycin and assessed neuronal death at 24 h after insult. OGD elicited neuronal death, as assessed by propidium iodide uptake (marker of degenerating neurons), and LDH release. Rapamycin did not detectably alter the number of propidium iodide (+) cells or the quantity of LDH released under normoxic conditions, but attenuated OGD-induced neuronal death (Figures 6a–c). While modest, the attenuation of neuronal death was significant in that protection of even a modest fraction of CA1 neurons is sufficient to preserve the functional integrity of CA1 neurons, assessed by electrophysiology41 or behavior.42

Pharmacological inhibition of mTORC1 or RNAi-mediated depletion of mTOR attenuates OGD-induced neuronal death. (a) Representative bright-field and fluorescent images of cultured hippocampal neurons DIV14 subjected to OGD or normoxia in the absence or presence of the mTORC1 inhibitor rapamycin for 24 h and stained with propidium iodide (PI) to mark dying cells. (b) Summary data of PI-stained neurons (OGD+vehicle: 347.6% increase from control; OGD+rapamycin: 263.1% increase from control); n=16–20 coverslips per treatment group/2 independent experiments. Bottom: (c) Summary data of lactate dehydrogenase (LDH) cytotoxicity assay, which measures release of LDH into the media, for hippocampal neurons subjected to OGD or normoxia in the presence or absence of rapamycin (OGD+vehicle: 179.1% increase from control; OGD+rapamycin: 158.2% increase from control); n=12 coverslips per treatment group in three independent experiments. (d) Representative bright-field and fluorescence images of cultured hippocampal neurons DIV14 subjected to OGD or normoxia in the absence or presence of mTOR or NT siRNA and stained with PI to mark dying cells. (e) Summary data of PI-stained neurons; n=16–20 coverslips per treatment group/2 independent experiments. (f) Summary data of LDH cytotoxicity assay in hippocampal neurons subjected to OGD or normoxia in the absence or presence of mTOR siRNA or NT siRNA. Data are normalized to control treatment; n=12 coverslips per treatment group in three independent experiments. Scale bar=50 μm. ***P<0.001, ##P<0.01, and #P<0.05.
Next, we examined the impact of si-mTOR on ischemia-induced neuronal death. Toward this end, we transfected hippocampal neurons with si-mTOR or NT siRNA before induction of OGD. si-mTOR, but not NT siRNA, attenuated OGD-induced neuronal death, assessed by the number of propidium iodide (+) cells (Figure 6d). Thus, inhibition or knockdown of mTOR before ischemia afforded partial rescue of neuronal death, consistent with an inverse causal relation between mTOR and autophagy or neuroprotection.
Inhibition of autophagy promotes ischemia-induced neuronal death
We next examined the impact of 3-methyladenine (3-MA) and C43, two well-established inhibitors of autophagy, on ischemia-induced neuronal death. 3-MA is an inhibitor of class III phosphatidylinositol 3-kinase and attenuates autophagy by blocking autophagosome formation.43, 44 Application of 3-MA or C43 to hippocampal neurons had little or no effect under normoxic conditions, but markedly decreased ischemia-induced neuronal death (Figures 7a–c). Consistent with this, siRNA to Atg7 (but not NT siRNA) markedly increased neuronal death (Figures 7d and e). Collectively, these findings strongly suggest that the protective effect afforded by mTOR inhibition requires autophagy.

Pharmacological inhibition of macroautophagy and RNAi-mediated depletion of Atg7 enhances OGD-induced neuronal death. (a) Representative bright-field and fluorescent images of cultured hippocampal neurons at DIV14 subjected to OGD or normoxia in the absence or presence of the autophagy inhibitor 3-MA or C43 for 24 h and stained with propidium iodide (PI) to mark dying cells. (b) Summary data of PI-stained neurons (OGD+vehicle: 324.3% increase from control; OGD+3-MA: 421.2% increase from control; OGD+C43: 660.3% increase from control); n=6–12 coverslips per treatment group/3 independent experiments. (c) Summary data of lactate dehydrogenase (LDH) cytotoxicity assay, which measures release of LDH into the media, for hippocampal neurons subjected to OGD or normoxia in the presence or absence of autophagy inhibitors. Left, OGD+vehicle: 207.6% increase from control; OGD+3-MA: 263.2% increase from control. Right, OGD+vehicle: 168.5% increase from control; OGD+C43: 205.8% increase from control); n=6–12 coverslips per treatment group/3 independent experiments. (d) Representative bright-field and fluorescence images of cultured hippocampal neurons at DIV14 subjected to OGD or normoxia in the absence or presence of siRNA to Atg7 or NT siRNA and stained with PI to mark dying cells. (e) Summary data of PI-stained neurons; n=6–12 coverslips per treatment group in three independent experiments. Scale bar=20 μm. ****P<0.0001, ***P<0.001, **P<0.01, ###P<0.001, and #P<0.05
Discussion
mTOR is a key regulator of cell growth, autophagy, translation, and survival. In neurons, components of the mTORC1 signaling cascade are strategically localized at pre- and postsynaptic sites where they influence synaptic plasticity by regulating the balance between protein synthesis and degradation. However, a role for mTORC1 signaling in neuronal death is not well delineated. In the present study, we show that global ischemia induces a transient increase in mTOR phosphorylation at S2448, but suppresses mTOR abundance and functional activity in selectively vulnerable CA1 neurons. The decrease in mTOR precedes or coincides with an increase in markers of autophagy, pS317-ULK-1, pS14-Beclin-1, and LC3-II, a decrease in the cargo adaptor p62, and an increase in autophagic flux. Concomitantly, ischemia phosphorylates and activates AMPK, which phosphorylates ULK-1 at S317, and acts in concert with loss of mTOR to promote induction of autophagy. This is significant in that autophagy, a programmed catabolic process downstream of mTORC1, promotes neuronal survival by targeting long-lived and misfolded proteins and damaged organelles to the lysosome for degradation.45 Inhibitors of the lysosomal (but not proteasomal) pathway rescue the ischemia-induced decrease in mTOR. Moreover, inhibition of autophagy or siRNA-mediated depletion of Atg7, a key component of autophagy, diminishes mTOR degradation, consistent with regulation of mTOR abundance via the autophagy/lysosomal pathway. Thus, mTOR is not only a negative regulator but also a target of autophagy-lysosome in insulted hippocampal neurons. Collectively, these findings identify a novel and previously unappreciated mechanism whereby mTOR regulates its own demise in a self-perpetuating manner in a clinically relevant model of global ischemia.
We further show that inhibition of mTOR by rapamycin or siRNA-mediated depletion of mTOR dephosphorylates and inactivates its downstream target p-757-ULK-1, releasing ULK-1 from mTOR and enabling it to associate with AMPK, which phosphorylates ULK-1 at S757 and initiates autophagy. This finding strengthens the conclusion that a decrease in mTOR abundance is sufficient to promote the initiation of autophagy. Our finding that increased mTOR phosphorylation at early times after ischemia correlates with a decrease of mTOR downstream targets is surprising. A possible scenario is that mTOR in neurons subjected to ischemia is divided into two pools, phosphorylated and non-phosphorylated mTOR. Phosphorylated mTOR is rapidly degraded. The remaining mTOR is not phosphorylated and cannot maintain phosphorylation of ULK-1 at S757. ULK-1 is released and associates with AMPK, which phosphorylates ULK-1 at S317 and initiates autophagy.32, 35 Alternatively, an endogenous inhibitor, such as DEPTOR,46 is induced in response to ischemia and binds p-mTOR, squelching its activity.
We propose a model whereby loss of mTOR promotes autophagy and attenuates neuronal death in a clinically relevant model of global ischemia (Figure 8). Under physiological conditions, mTOR serves as a brake on autophagy.11, 12, 13 Induction of ischemia triggers a transient increase in mTOR phosphorylation, while decreasing mTOR abundance and functional activity in hippocampal neurons destined to die. At the same time, ischemia activates AMPK, which acts in concert with loss of mTOR to promote induction of autophagy. The decrease in mTOR removes the brake normally imposed on autophagy; upon release, autophagy degrades mTOR via the autophagy/lysosomal pathway, resulting in a self-perpetuating loss of mTOR. Thus, a decrease in mTOR abundance, as well as activity, can activate downstream targets that converge on autophagy. Moreover, inhibition of autophagy by C43- or siRNA-mediated depletion of Atg7 promotes autophagy and reduces neuronal death. Thus, autophagy promotes neuroprotection, representing a ‘failed attempt’ of the cell to survive in the face of ischemia.

Model depicting a hypothetical mechanism by which mTOR self-regulates its own abundance via the lysosome/autophagy pathway following global ischemia. Left: Under physiological conditions, mTOR imposes a brake on autophagy by phosphorylation and inactivation of ULK-1 at S757, retaining ULK-1 away from AMPK, thereby imposing a break on autophagy. Right: In response to ischemia, mTOR phosphorylation/inactivation of ULK-1 is decreased, whereas AMPK-dependent phosphorylation/activation of ULK-1 is increased. This, in turn, removes the brake on initiation of autophagy and promotes ULK-1-mediated phosphorylation of Beclin-1 at S14, which drives induction of autophagy. The increase in autophagy further degrades mTOR via the lysosome, providing a feedback mechanism by which mTOR self-regulates its own abundance in CA1 neurons destined to die
Our findings do not, however, preclude the possibility that other upstream and/or downstream regulators of mTORC1 could also impact on autophagy. Of note, recent findings of Buchan and co-workers47, 48 reveal that ischemia induces expression of hamartin, an upstream inhibitor of mTOR, in CA3 neurons and that hamartin is causally related to the resistance of CA3 neurons to ischemia-induced cell death. In contrast, we find that ischemia suppresses mTOR and activates autophagy in CA1 neurons destined to die. Our results represent an advance over previous studies in that we delineate mechanisms underlying ischemia-induced activation of autophagy and reveal a novel mechanism by which mTOR self-regulates its own degradation in insulted hippocampal neurons. Although not addressed by the present study, it is likely that Akt, which is activated by ischemia,49 activates p-mTOR at S2448. Alternatively, given that pS6 is also activated by ischemia (not illustrated), S6 kinase might activate mTOR.
In response to cellular stress, mTOR translocates to the lysosomal surface in close proximity to upstream effectors that phosphorylate mTOR at S2448.15, 16, 50 In cancer cells and fibroblasts, translocation of mTOR to the lysosome is dependent on assembly with the vacuolar (H+) ATPase, which is situated on the lysosomal membrane,51 and members of the Rag family of proteins that bind to the mTOR complex and shuttle it to the lysosomal surface.15, 52 A novel finding in the present study is that ischemic insults not only target mTOR to lysosomes but also induce its degradation. These results identify mTOR degradation as a novel mechanism to inactivate mTOR at the lysosomal membrane under conditions of cellular stress, a mechanism relevant to achieving maximal activation of autophagy in neurons. However, the mechanism by which mTOR translocates to the lysosome in insulted neurons is, as yet, unknown.
mTOR activity is tightly regulated by phosphorylation at S2448,10, 53 and by assembly with kinase-associated proteins in neuronal and non-neuronal cells.1, 30, 54 The present study represents an advance over previous studies in that we show that ischemia transiently increases the phosphorylation status of mTOR at S2448, while decreasing its abundance and functional activity, as assessed by the phosphorylation status and activation state of two prominent downstream targets, 4E-BP and ULK-1. Thus, the phosphorylation status of mTOR does not necessarily predict its kinase activity. Consistent with this notion, in cancer cells, mutation of mTOR at S2448 to alanine does not alter the ability of mTOR to regulate downstream targets.55 These findings highlight the complexity of signaling pathways upstream and downstream of mTOR.
In summary, we show that global ischemia promotes mTOR degradation and activation of autophagy in neurons before neuronal death. We further show an inverse causal relation between mTOR and autophagy and/or neuronal death. In addition, we show that the ischemia-induced decrease in mTOR occurs via lysosomal degradation. To our knowledge, our findings are the first to demonstrate that mTOR is not only a negative regulator but also a target of the autophagy-lysosome system in insulted hippocampal neurons. Collectively, our findings support a role for mTORC1-dependent autophagy as a novel therapeutic target for intervention in ischemic injury and potentially other brain disorders.
Materials and Methods
Animals
Age-matched adult male Sprague–Dawley rats (Charles River Laboratories, Wilmington, MA, USA) weighing 150–200 g were maintained in a temperature- and light-controlled environment with a 12:12 h light–dark cycle and were treated in accordance with the principles and procedures of the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Protocols were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine (New York, NY, USA).
Global ischemia
Animals were subjected to transient global ischemia by the four-vessel occlusion paradigm described previously.38, 49, 56, 57 For sham surgery, animals were subjected to the same anesthesia and surgical conditions, except that the vertebral and carotid arteries were not occluded. Body temperature was maintained at 37.5±0.5 °C by external warming until recovery from anesthesia. Animals that failed to show complete loss of the righting reflex and dilation of the pupils 2 min after occlusion, as well as animals that exhibited abnormal vocalization, generalized convulsions, or hypoactivity were excluded from the study.
Western blotting
Western blot analysis was performed as described previously.49, 56, 57 In brief, cells were washed with cold 1 × PBS and lysed in buffer containing the following (in mM): 50 Tris-HCl, pH 7.5, 150 NaCl, 1% Igepal CA-630, 1% sodium deoxycholate, 0.1% SDS supplemented with 1 × protease inhibitor mixture (Sigma, St. Louis, MO, USA). Cell lysates were centrifuged (10 min, 14 000 r.p.m.) to separate soluble and insoluble fraction. Insoluble fractions were were lysed and sonicated in the same lysis buffer with 2% SDS.
For tissue samples, hippocampi were rapidly dissected and transverse slices of dorsal hippocampus (1 mm) were cut with a McIlwain tissue chopper. The CA1 and CA3 subfields were rapidly microdissected and homogenized in buffer containing the following (in mM): 20 HEPES, pH 7.4, 10 KCl, 10 MgCl2, PMSF (17 μg/ml), supplemented with 1 × protease inhibitor mixture. Homogenate was centrifuged at a low speed (3000 × g, 10 min, 4 °C) and the resulting pellet was rehomogenized and centrifuged at a high speed (10 000 × g, 15 min, 4 °C). The resulting supernatant was removed and lysed in 1:1 volume of lysis buffer containing the following (in mM): 50 Tris-HCl, pH 7.5, 250 NaCl, 5 EDTA, 0.25% sodium deoxycholate, 0.1% SDS, 0.1% Triton supplemented with 1 × protease inhibitor mixture. The resulting pellet, termed the insoluble fraction, was lysed and sonicated in the same lysis buffer with 2% SDS. Protein concentrations were measured with the bicinchoninic acid assay (Pierce, Thermo Fisher Scientific, Waltham, MA, USA). Equal aliquots of protein (20–40 μg) were subjected to SDS-PAGE (4–20%), transferred to a nitrocellulose or PVDF membrane, and processed for incubation with primary and secondary HRP-conjugated antibodies. After incubations, membranes were washed and reacted with enhanced chemiluminescence reagent (GE Healthcare Life Sciences, Pittsburgh, PA, USA).
qRT-PCR
For tissue samples, the CA1 was rapidly microdissected from transverse slices of dorsal hippocampus cut by a McIlwain tissue chopper (1 mm). RNA was extracted using mirVana (Ambion, Thermo Fisher Scientific, Waltham, MA, USA), and concentration was measured with a Nanodrop (Thermo Fisher Scientific). Aliquots of RNA (10 ng) were reverse transcribed to cDNA with the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). qRT-PCR (qPCR) was performed using TaqMan probes (Applied Biosystems) for mTOR (reference number: Rn00693900_m1); hypoxanthine phospho-ribosyl transferase (Reference Rn01318745_g1), which is invariant in its expression following global ischemia,58 served as an endogenous reference. Reactions were run in triplicate in a StepOnePlus Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific). The relative change in the mRNA expression was determined by the equation: fold change=2−ΔCt (ΔCt=Ct target−Ct reference). The Ct value corresponds to the cycle number at which the fluorescence signal crosses a threshold. Relative expression ratios were calculated by normalization of values for experimental samples to those of the corresponding sham controls.
Immunohistochemistry
Immunohistochemistry was performed on sections of rat brain 24 h after ischemia as described.36, 38, 49 Briefly, rats were killed under deep anesthesia by transcardial perfusion with 4% paraformaldehyde in PBS. Brains were immersion fixed in 4% paraformaldehyde in PBS overnight, washed with 30% sucrose in phosphate buffer solution, and frozen with 2-methylbutane. Frozen brains were cryostat sectioned at 20 μm. Sections were blocked and incubated for 48 h at 4 °C with antibody to mTOR and for 1 h with antibody to NeuN, followed by incubation with goat secondary antibodies Alexa 488 and Alexa 543 (1:500; Molecular Probes, Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at room temperature. The specificity of immunolabeling was confirmed by incubation of sections with preimmune rabbit IgG in place of primary antibody; under these conditions, no detectable labeling was observed. Sections were viewed and images were acquired through a Zeiss DUO V2 laser scanning confocal fluorescence microscope (Carl Zeiss Microscopy, LLC, Thornwood, NY, USA). Images were processed with ImageJ software (Public Domain, imagej.nih.gov/ij/).
Drug injections in vivo
Leupeptin (10 mM) was dissolved in DMSO or DMSO was administered to animals immediately after ischemia or sham surgery by intracerebroventricular injection. For intracerebroventricular injection, a 28-gauge needle attached to a Hamilton syringe was stereotaxically lowered into the right lateral ventricle to a position defined by the following coordinates relative to bregma: 0.92 mm posterior, 1.2 mm lateral, and 3.6 mm below the skull surface. A flow rate of 1 μl/min (total 5 μl) was used.
Primary cultures of hippocampal neurons and OGD
Primary cultures of hippocampal neurons were prepared from embryonic day 18 (E18) rats as described previously.38, 56 Cultures were maintained 14 days in vitro before experiments. To induce OGD, cultures were exposed to serum-free, glucose-free artificial cerebrospinal fluid (aCSF) saturated with 95%N2/5%CO2 (30 min at 37 °C) in an airtight, anoxic chamber (Billups-Rothenberg, San Diego, CA, USA). Cultures were returned to oxygenated, glucose-containing medium under normoxic conditions in the presence or absence of indicated drugs for 3 h. After incubation, cells were returned to normal medium and harvested at indicated times for western blot analysis or treated with propidium iodide (1:20 000) at 24 h after OGD to assess neuronal death. For oxygen deprivation alone (hypoxia), hippocampal neurons were exposed to normal medium with serum saturated with 95%N2/5%CO2 (30 min at 37 °C) in an airtight chamber. For glucose deprivation alone, hippocampal neurons were exposed to serum-free, glucose-free aCSF in normal, oxygen-containing incubation conditions (30 min at 37 °C).
Cell culture, drug treatment, and transfection
N2A cells were plated on culture dishes or plates incubated in DMEM with 10% fetal bovine serum with an atmosphere of 10% CO2 at 37 °C. Cells were grown to 80–90% confluency for drug treatments and 40–50% for transfection. Cells were transfected with myc-mTOR plasmid using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Drugs treated at indicated concentrations included: clasto-lactacystin β-lactone (Lactacystin) (Millipore, Billerica, MA, USA), ammonium chloride (Sigma), leupeptin (Millipore), rapamycin (Sigma), cycloheximide (Millipore), staurosporine (Millipore), and hydrogen peroxide (Sigma).
siRNA oligonucleotides
siRNA oligonucleotides targeting endogenous rat mTOR, Atg7, and non-targeting constructs, which target no known vertebrate gene, were obtained through Dharmacon Accell RNAi Libraries (GE Healthcare, Dharmacon, Lafayette, CO, USA). siRNA constructs were diluted in buffer (GE Healthcare) and transfected in culture medium at a working concentration of 1 μM. The efficacy of mTOR or Atg7 siRNA was evaluated by western blot 4 days after transfection.
Lactate dehydrogenase assay
To quantify the amount of LDH released into the medium, we incubated 50 μl of cell culture medium from each sample well in triplicate with 100 μl of LDH Reaction Mix from the LDH Cytotoxicity Assay Kit II (Abcam, Cambridge, UK) for 30 min at room temperature. Fifty microliters of normal culture medium was used as a background control. LDH readings were taken on a 96-well plate reader with an absorbance wavelength of 450 nm. LDH cytotoxicity values were calculated by subtracting the background control from the reading from each sample well.
Antibodies
Antibodies used in this study include: anti-mTOR, 1:1000; anti-p-mTOR (S2448), 1:2000; anti-p-4E-BP, 1:1000; anti-4E-BP, 1:500; anti-p-ULK (S757), 1:1000; anti-p-ULK (S317), 1:1000; anti-ULK, 1:1000; anti-Beclin-1, 1:500; anti-p62, 1:1000; anti-p-p70S6K (T389), 1:500; anti-p70S6K, 1:1000; anti-AMPK, 1:1000; anti-ubiquitin, 1:1000 (Cell Signaling Technology, Danvers, MA, USA); anti-mTOR, 1:100 (Abcam); anti-NeuN, 1:1000 (Millipore); anti-p-Beclin-1 (S14), 1:500 (Abbiotec, San Diego, CA, USA); anti-LC3-I/II, 1:2000, anti-β-actin, 1:5000 (Sigma); anti-GAPDH, 1:5000, (Fitzgerald, Acton, MA, USA).
Data analysis
One-way analysis of variance was used for all statistical analysis with multiple comparisons. For all P-values, the Student’s t-test was used for post hoc analysis. Results are reported as mean±S.E.M. Significance was taken as P<0.05.
References
Hay N, Sonenberg N . Upstream and downstream of mTOR. Genes Dev 2004; 18: 1926–1945.
Klann E, Dever TE . Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci 2004; 5: 931–942.
Laplante M, Sabatini DM . mTOR signaling in growth control and disease. Cell 2012; 149: 274–293.
Huang J, Manning BD . A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans 2009; 37: 217–222.
Darnell JC, Klann E . The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci 2013; 16: 1530–1536.
Huber KM, Klann E, Costa-Mattioli M, Zukin RS . Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J Neurosci 2015; 35: 13836–13842.
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36: 585–595.
Hoeffer CA, Klann E . mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci 2010; 33: 67–75.
Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N . Translational control of long-lasting synaptic plasticity and memory. Neuron 2009; 61: 10–26.
Zhang Y, Nicholatos J, Dreier JR, Ricoult SJ, Widenmaier SB, Hotamisligil GS et al. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 2014; 513: 440–443.
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20: 1992–2003.
Rubinsztein DC, Codogno P, Levine B . Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11: 709–730.
Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15: 741–750.
Wong E, Cuervo AM . Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 2010; 13: 805–811.
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM . Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010; 141: 290–303.
Avruch J, Hara K, Lin Y, Liu M, Long X, Ortiz-Vega S et al. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene 2006; 25: 6361–6372.
Chiang GG, Abraham RT . Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem 2005; 280: 25485–25490.
Holz MK, Blenis J . Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem 2005; 280: 26089–26093.
Harris H, Rubinsztein DC . Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 2012; 8: 108–117.
Dann SG, Selvaraj A, Thomas G . mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med 2007; 13: 252–259.
Sabatini DM . mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 2006; 6: 729–734.
Sarbassov DD, Ali SM, Sabatini DM . Growing roles for the mTOR pathway. Curr Opin Cell Biol 2005; 17: 596–603.
Sawicka K, Zukin RS . Dysregulation of mTOR signaling in neuropsychiatric disorders: therapeutic implications. Neuropsychopharmacology 2012; 37: 305–306.
Lo EH, Dalkara T, Moskowitz MA . Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 2003; 4: 399–415.
Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H et al. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal 2011; 14: 1505–1517.
Moskowitz MA, Lo EH, Iadecola C . The science of stroke: mechanisms in search of treatments. Neuron 2010; 67: 181–198.
Ofengeim M, Miyawaki T, Zukin RS Molecular and cellular mechanisms of ischemia-induced neuronal death. In: Mohr JP, Choi DW, Grotta JC, Weir B, Wolf PA et al. (eds). Stroke: Pathophysiology, Diagnosis and Management. Churchill Livingstone Elsevier: Philadelphia, PA, 2011, pp 1–39.
Vosler PS, Gao Y, Brennan CS, Yanagiya A, Gan Y, Cao G et al. Ischemia-induced calpain activation causes eukaryotic (translation) initiation factor 4G1 (eIF4GI) degradation, protein synthesis inhibition, and neuronal death. Proc Natl Acad Sci USA 2011; 108: 18102–18107.
Tanaka H, Yokota H, Jover T, Cappuccio I, Calderone A, Simionescu M et al. Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J Neurosci 2004; 24: 2750–2759.
Laplante M, Sabatini DM . Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci 2013; 126: 1713–1719.
Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM . A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012; 485: 109–113.
Kim J, Kundu M, Viollet B, Guan KL . AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13: 132–141.
Anjum R, Blenis J . The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol 2008; 9: 747–758.
Hardie DG, Ross FA, Hawley SA . AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012; 13: 251–262.
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331: 456–461.
Calderone A, Jover T, Noh K-M, Tanaka H, Yokota H, Lin Y et al. Ischemic insults de-repress the gene silencer rest in neurons destined to die. J Neurosci 2003; 23: 2112–2121.
Noh KM, Yokota H, Mashiko T, Castillo PE, Zukin RS, Bennett MV . Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc Natl Acad Sci USA 2005; 102: 12230–12235.
Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM et al. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc Natl Acad Sci USA 2012; 109: E962–E971.
Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E . Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 2006; 51: 441–454.
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169: 425–434.
Takeuchi K, Yang Y, Takayasu Y, Gertner M, Hwang JY, Aromolaran K et al. Estradiol pretreatment ameliorates impaired synaptic plasticity at synapses of insulted CA1 neurons after transient global ischemia. Brain Res 2015; 1621: 222–230.
Gulinello M, Lebesgue D, Jover-Mengual T, Zukin RS, Etgen AM . Acute and chronic estradiol treatments reduce memory deficits induced by transient global ischemia in female rats. Horm Behav 2006; 49: 246–260.
Seglen PO, Gordon PB . 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 1982; 79: 1889–1892.
Takatsuka C, Inoue Y, Matsuoka K, Moriyasu Y . 3-Methyladenine inhibits autophagy in tobacco culture cells under sucrose starvation conditions. Plant Cell Physiol 2004; 45: 265–274.
Cuervo AM, Wong E . Chaperone-mediated autophagy: roles in disease and aging. Cell Res 2014; 24: 92–104.
Zhao Y, Xiong X, Sun Y . DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell 2011; 44: 304–316.
Das G, Shravage BV, Baehrecke EH . Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol 2012; 4: a008813.
Papadakis M, Hadley G, Xilouri M, Hoyte LC, Nagel S, McMenamin MM et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat Med 2013; 19: 351–357.
Miyawaki T, Ofengeim D, Noh KM, Latuszek-Barrantes A, Hemmings BA, Follenzi A et al. The endogenous inhibitor of Akt, CTMP, is critical to ischemia-induced neuronal death. Nat Neurosci 2009; 12: 618–626.
Inoki K, Li Y, Xu T, Guan KL . Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003; 17: 1829–1834.
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM . mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011; 334: 678–683.
Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM . Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012; 150: 1196–1208.
Pearce LR, Komander D, Alessi DR . The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 2010; 11: 9–22.
Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009; 137: 873–886.
Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM et al. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res 2000; 60: 3504–3513.
Kaneko N, Hwang JY, Gertner M, Pontarelli F, Zukin RS . Casein kinase 1 suppresses activation of REST in insulted hippocampal neurons and halts ischemia-induced neuronal death. J Neurosci 2014; 34: 6030–6039.
Sehara Y, Sawicka K, Hwang JY, Latuszek-Barrantes A, Etgen AM, Zukin RS . Survivin is a transcriptional target of STAT3 critical to estradiol neuroprotection in global ischemia. J Neurosci 2013; 33: 12364–12374.
Formisano L, Noh KM, Miyawaki T, Mashiko T, Bennett MV, Zukin RS . Ischemic insults promote epigenetic reprogramming of mu opioid receptor expression in hippocampal neurons. Proc Natl Acad Sci USA 2007; 104: 4170–4175.
Acknowledgements
We thank Drs Ana Maria Cuervo, Helin Norberg, and Kelly Aromolaran for their critical evaluation of the manuscript. We also thank Dr Matt Micsenyi for helpful scientific discussions and intellectual guidance. This work was supported by National Institutes of Health Grant Nos. NS46742 and MH092877, and the generous grant from the FM Kirby Foundation (to RSZ), AHA Scientist Development Grant No. 16SDG31500001, and NARSAD Young Investigator Grant 25369 (to J-YH). RSZ is the FM Kirby Professor in Neural Repair and Protection.
Author contributions
J-YH, MJG, DO and RSZ designed the research; J-YH, MJG, FP, BC-V and DO performed experiments; J-YH, MJG, DO, MVLB and RSZ analyzed the data; J-YH, MJG, DO, MVLB and RSZ wrote the paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Freeman
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Hwang, JY., Gertner, M., Pontarelli, F. et al. Global ischemia induces lysosomal-mediated degradation of mTOR and activation of autophagy in hippocampal neurons destined to die. Cell Death Differ 24, 317–329 (2017). https://doi.org/10.1038/cdd.2016.140
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.140
This article is cited by
-
Neuroprotective mechanisms of luteolin in glutamate-induced oxidative stress and autophagy-mediated neuronal cell death
Scientific Reports (2024)
-
Rapamycin ameliorates brain damage and maintains mitochondrial dynamic balance in diabetic rats subjected to middle cerebral artery occlusion
Metabolic Brain Disease (2023)
-
Reinventing the Penumbra — the Emerging Clockwork of a Multi-modal Mechanistic Paradigm
Translational Stroke Research (2023)
-
The Pathological Mechanism of Neuronal Autophagy-Lysosome Dysfunction After Ischemic Stroke
Cellular and Molecular Neurobiology (2023)
-
Novel mechanism of napabucasin, a naturally derived furanonaphthoquinone: apoptosis and autophagy induction in lung cancer cells through direct targeting on Akt/mTOR proteins
BMC Complementary Medicine and Therapies (2022)
