
Abstract
IκB kinase β (IKKβ) is a catalytic subunit of the IKK complex, which activates nuclear factor-κB (NF-κB). Although its role in osteoclastogenesis is well established, the role of IKKβ in bone formation is poorly understood. Here, we report that conditional knockout of Ikkβ in limb bud mesenchymal cells results in the upregulation of monocyte chemoattractant protein-5 (MCP-5) in the perichondrium, which in turn inhibits the growth of longitudinal bone by compromising chondrocyte hypertrophy and increasing the apoptosis of chondrocytes within the growth plate. Contrary to expectations, IKKβ in cells of chondrocyte or osteoblast lineage was dispensable for bone growth. On the other hand, ex vivo experiments confirmed the role of MCP-5 in the growth of longitudinal bone. Furthermore, an in vitro study demonstrated that the action of IKKβ on MCP-5 is cell autonomous. Collectively, our results provide evidence for a previously unrecognized role of IKKβ in the regulation of the growth plate that is mediated through stimulation-independent downregulation of MCP-5 in the perichondrium.
Similar content being viewed by others


Main
The IκB kinase (IKK) complex contains two catalytic subunits, IKKα and IKKβ, which are highly similar in sequence.1 In conjunction with the regulatory subunit, IKKγ (also known as NEMO), the IKK complex transduces signals to downstream effectors.2 Among the downstream targets of the IKK complex, nuclear factor-κB (NF-κB) is known to have a plethora of roles in immune and inflammatory response, cell growth, cell death and stress responses.3, 4 In addition, recently, it has been reported that IKKβ targets additional substrates and regulates NF-κB-independent signaling.5, 6 Because mice deficient for the gene encoding IKKβ die in midgestation,7, 8, 9 to investigate the function of IKKβ in vivo in tissue- and stage-specific settings, mice with a floxed Ikkβ allele have been generated.10, 11
The skeleton is an organ that not only supports and protects the body, but is also involved in other functions via communications with other organs. These communications render unanticipated complexities to skeletal patterning and to the specification/differentiation of skeletal cells during development.12 The skeleton is composed of cartilage and bone, and the development of these two cell types is coordinated by a network of signaling pathways and transcription factors.13 Bone is remodeled by the coupling of two opposing processes: bone resorption and bone formation. During bone resorption, osteoclasts degrade mature bone tissue, whereas during bone formation, osteoblasts form new bone through a process called ossification. The development and activation of osteoclasts are well characterized at the genetic and molecular levels,14, 15 and the role of IKK signaling in these processes has been established.16 In contrast, less is known about the role of IKK in osteoblast development.
In this study, we sought to explore the role of IKKβ in bone formation through osteoblast- or chondrocyte-specific ablation of Ikkβ. Although IKKβ was dispensable for cells of either osteoblast or chondrocyte lineage, loss of IKKβ in limb bud mesenchymal cells resulted in the growth retardation of longitudinal bone. This effect was due to a reduced hypertrophy and increased apoptosis of chondrocytes in the growth plate. A search for the mechanism underlying this abnormality led to the finding that IKKβ suppresses the expression of Mcp-5 (monocyte chemoattractant protein-5) in the perichondrium in a cell-autonomous manner. Based on these results, we suggest that the IKKβ–MCP-5 axis may regulate chondrocyte hypertrophy in parallel with the transforming growth factor-β receptor type II (TβRII)–MCP-5 axis, which was reported recently.17
Results
Loss of IKKβ in the osteoblast lineage does not affect bone remodeling
Bone remodeling depends on the orchestrated balance between bone formation and bone resorption. The role for IKK–NF-κB signaling in osteoclastogenesis is well established,16 but far less is known about role for IKKβ in bone formation. We therefore crossed IkkβF/F mice10 with mice expressing Cre recombinase under the control of type I collagen promoter elements.18 In the resulting IkkβF/F;Col1a1-Cre (hereafter described as IkkβCol1KO) mice, deletion of the Ikkβ locus should occur specifically in mesenchymal bone cells through Cre recombination and can be assessed by PCR preferentially amplifying IkkβΔ DNA (Supplementary Figure S1a).19 Indeed, as shown in Supplementary Figures S1b and c, the IkkβΔ fragment was amplified only from genomic DNA prepared from the bone of IkkβCol1KO mice, demonstrating the specificity of the recombination in bone. To quantify the efficiency of the deletion more accurately, we performed qPCR with a primer set that only amplifies the residual Ikkβ locus19 and observed efficient (78.5%) deletion of the Ikkβ locus in primary osteoblasts of IkkβCol1KO mice (Figure 1a and Supplementary Figure S1a). Despite the efficient deletion of Ikkβ in osteoblasts, IkkβCol1KO mice were born at the expected Mendelian frequency, and the length of their crown–rump and long bones were the same as those of control mice until 8 weeks of age (Supplementary Figure S2). We next assessed the skeletal volume of IkkβCol1KO mice in detail using microcomputed tomography (μCT) (Figure 1b). μCT analysis of the secondary spongiosa of the distal femur revealed that bone volume in the cancellous and cortical bones of IkkβCol1KO mice was not significantly different from that of control mice at age 4 weeks and even 8 weeks (Figures 1c and d). We further performed dynamic histomorphometric analysis by labeling the bones with calcein, a well-known marker of bone formation, over 7 days. The bone formation rate of 8-week-old IkkβCol1KO mice (77.49 μm3/μm2 per year) was similar to that of control mice (78.14 μm3/μm2 per year) (Figure 1e), as too was the number of osteoblasts and osteoclasts (Figures 1f and g). These results indicate that IKKβ in cells of osteoblast lineage is dispensable for normal bone growth.

IKKβ in osteoblast lineage is dispensable for bone growth and remodeling during normal development. (a) The effect of deleting the Ikkβ locus was examined using genomic DNA isolated from primary osteoblasts of the indicated mouse. Residual Ikkβ locus was quantified by qPCR. (b) μCT images of the distal femur from 4-week-old IkkβF/F (control) and IkkβF/F;Col1a1-Cre mice. (c) Cancellous BV/TV in the distal femur of 4- and 8-week-old control and IkkβF/F;Col1a1-Cre mice (n=10 in each group). (d) Cortical BV/TV in the distal femur of 4- and 8-week-old control and IkkβF/F;Col1a1-Cre mice (n=10 in each group). (e) Bone formation rate was calculated with calcein double labeling (n=10 in each group). (f and g) Osteoblast and osteoclast numbers of 8-week-old control and IkkβF/F;Col1a1-Cre mice. *P<0.001, two-sided Student’s t-test. Error bars±S.D.
Loss of IKKβ in osteoblast lineage does not affect bone loss induced by ovariectomy
As there was no abnormality in IkkβCol1KO mice at physiological conditions, we next investigated the role of IKKβ in osteoblast lineage during bone loss induced by ovariectomy. To mimic the bone loss in postmenopausal osteoporosis in humans, the ovariectomy mouse model has been widely used.20 We performed sham operation or ovariectomy to 12-week-old control and IkkβCol1KO mice and analyzed them 8 weeks later. μCT analysis revealed that cancellous bones in the distal femur were significantly decreased by ovariectomy compared with those of sham operated in both control and IkkβCol1KO mice (Figure 2a). Yet, there was no significant difference in bone volume per trabecular volume (BV/TV) of ovariectomized IkkβCol1KO mice compared with that of control ovariectomized mice (Figure 2b). These results in conjunction with those presented in Figure 1 indicate that IKKβ in cells of osteoblast lineage is dispensable not only under physiological conditions but also during postmenopausal osteoporosis.

IKKβ in osteoblast is dispensable for cancellous bone loss induced by ovariectomy. (a) μCT images of the distal femur from adult control and IkkβF/F;Col1a1-Cre mice after ovariectomy (OVX). (b) Cancellous BV/TV in the distal femur of sham operated (IkkβF/F sham) and ovariectomized (IkkβF/F OVX) control and IkkβF/F;Col1a1-Cre mice (n=6 in each group). *P<0.05, two-sided Student’s t-test. Error bars±S.D.
Loss of IKKβ in limb bud mesenchymal cells compromises postnatal longitudinal bone growth
To examine the role of IKKβ in endochondral ossification in vivo, we crossed the IkkβF/F mouse line with a transgenic mouse line expressing Cre recombinase under the control of Prx1 enhancer elements.21 Prx1-Cre transgenic mice express Cre recombinase in mesenchymal cells of the developing limb and parts of the skull but not in the spine or other organs. IkkβF/F;Prx1-Cre (hereafter described as IkkβPrx1KO) mice were born at the expected Mendelian frequency and were viable and fertile. We confirmed the depletion of Ikkβ mRNA in the growth plate of 2-week-old IkkβPrx1KO mice compared with control mice (Figure 3a). IkkβPrx1KO mice had no skeletal abnormality at birth (Figures 3b and c). Although no significant difference in the length of lumbar vertebrae (L2–5) was observed, the long bones were shorter than those of control mice at 2 weeks of age and the difference lasted even at 8 weeks (Figures 3d and e). These results indicate that IKKβ in the mesenchymal cells of developing limb bud, which are known to be the precursors of osteochondro progenitor cells, is involved in postnatal growth of the longitudinal bone.

Phenotype of mesenchymal cell-specific Ikkβ knockout mice. (a) Ikkβ mRNA level in the growth plate of control and IkkβF/F;Prx1-Cre (n=3 in each group; *P<0.001, two-sided Student’s t-test). Dissection of the growth plate and isolation of RNA was performed as described in the Materials and Methods section. (b) Skeletal preparation at P1 was stained with alcian blue and alizarin red. (c) Detailed skeletal morphology at P1. Lumber vertebrae (top panel), upper extremity (middle panel) and lower extremity (bottom panel). (d) Photograph of 8-week-old control and IkkβF/F;Prx1-Cre mice (left panel) and femoral bone (right panel). IkkβF/F;Prx1-Cre mice exhibited postnatal short long bones. (e) The length of lumber vertebrae (L2–5) and femur of control and IkkβF/F;Prx1-Cre mice from 1 day to 8 weeks. Although there was no significant difference in the length of lumbar vertebrae, the length of long bones was significantly shorter in IkkβF/F;Prx1-Cre mice by 2 weeks of age and persisted at 8 weeks (n=18 in control and n=20 in IkkβF/F;Prx1-Cre; *P<0.01, two-sided Student’s t-test). Error bars±S.D.
Loss of IKKβ in mesenchymal cells affects growth plate structure
We further investigated bone defects in IkkβPrx1KO mice. Histomorphometric analysis of proximal tibia from 2-week-old IkkβPrx1KO mice revealed a significant reduction in growth plate thickness owing to a reduction in thickness of the hypertrophic zone (Figures 4a and b). As shown in Figures 4c and d, we confirmed the reduction in the hypertrophic zone by immunohistochemical analysis of type X collagen. To further investigate the cause of chondrocyte abnormality at the growth plate of IkkβPrx1KO mice, cell proliferation and apoptosis were assessed by BrdU staining and terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL) staining, respectively. For this purpose, tibial sections prepared from 2-week-old mice were studied. BrdU labeling demonstrated that BrdU-positive proliferating chondrocytes in IkkβPrx1KO and control mice were not significantly different (Figures 4e and f). On the other hand, TUNEL staining demonstrated that chondrocyte apoptosis was significantly increased in the chondroosseus junction of IkkβPrx1KO mice compared with that of control mice (Figures 4g and h). These results indicate that the loss of IKKβ in mesenchymal cells of the limb bud inhibits chondrocyte hypertrophy through an increase in chondrocyte apoptosis in the growth plate.

Loss of IKKβ in limb bud mesenchymal cells alters the growth plate structure. (a) Proximal tibial growth plates from 2-week-old mice were stained with safranin-O. A reduction in thickness of the growth plate was observed in IkkβF/F;Prx1-Cre mice. (b) Histomophometric analysis of the growth plate. The thickness of total growth plate was significantly shorter than that of controls (n=8 in each group). (c) Tibiae from 2-week-old control and IkkβF/F;Prx1-Cre mice were immunostained for type X collagen. (d) Thickness of the hypertrophic zone in IkkβF/F;Prx1-Cre mice was significantly shorter than that of controls (n=5 in each group). (e) Two-week-old control and IkkβF/F;Prx1-Cre mice were injected with BrdU labeling solution (1 ml/100 g body weight) at 20 and 4 h before killing, and proximal tibiae were immunostained for BrdU. (f) BrdU-positive cells were quantitatively represented (n=5 in each group). (g) TUNEL assay in 2-week-old proximal tibiae from control and IkkβF/F;Prx1-Cre mice. Apoptotic cells are indicated (red arrows). (h) Proximal tibiae from 2-week-old IkkβF/F;Prx1-Cre mice showed increased chondrocyte apoptosis in the chondroosseous junction (n=5 in each group). *P<0.05 two-sided Student’s t-test. Error bars±S.D. r, resting zone; p, proliferative zone; h, hypertrophic zone
No significant difference in cancellous and cortical BV/TV was observed between IkkβPrx1KO mice and control mice at 4 and 8 weeks of age according to μCT analysis of the distal femur (Supplementary Figures S3a and b). The bone formation rate and the number of osteoblasts and osteoclasts in 8-week-old IkkβPrx1KO mice did not show significant differences compared with control mice (Supplementary Figures S3c, d and e). These results indicate that the phenotype of IkkβPrx1KO mice is due to the defect in hypertrophic chondrocytes in the growth plate but not in bone remodeling.
Loss of IKKβ in chondrocytes does not cause bone growth retardation
It is known that appendicular and axial skeletons develop through endochondral ossification in mammals, and, in the primitive stage of this process, two distinct cell types, chondrocytes and perichondral cells, appear.22 To dissect the contribution of the two cell types to the growth plate abnormalities in IkkβPrx1KO mouse, we crossed IkkβF/F mice with transgenic mice that express Cre recombinase under the control of a type II collagen promoter.23, 24 IkkβF/F;Col2a1-Cre (hereafter described as IkkβCol2KO) mice were born at the expected Mendelian frequency and were viable and fertile. The deletion efficiency of the IkkβF/F locus in IkkβCol2KO mice was assessed with genomic DNA isolated from primary rib chondrocytes by measuring the residual Ikkβ locus. As shown in Figure 5a, the amount of residual Ikkβ locus was 3.4% in primary chondrocytes isolated from IkkβCol2KO, demonstrating that the deletion is quite effective. We examined the length of lumbar vertebrae and long bones until 8 weeks of age, but IkkβCol2KO mice did not show any growth retardation compared with control mice (Figure 5b). These results indicate that IKKβ in cells committed to chondrocyte lineage is dispensable for normal bone growth in vivo.

IKKβ in chondrocytes is dispensable for endochondral ossification. (a) qPCR of residual Ikkβ exon 3 with genomic DNA isolated from primary chondrocytes of control and IkkβF/F;Col2a1-Cre mice (n=3 in each group) (b) Length of indicated bone of 8-week-old IkkβF/F;Col2a1-Cre mice (n=5). *P<0.001, two-sided Student’s t-test. Error bars±S.D.
Loss of IKKβ in the perichondrium causes aberrant expression of MCP-5
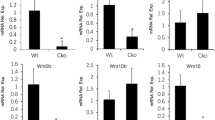
Reduced thickness of the hypertrophic zone in IkkβPrx1KO mice could be attributed to the loss of IKKβ in mesenchymal cells or their derivatives except for chondrocyte- and osteoblast-lineage cells. Cre recombinase has been reported to be expressed in the perichondrium from the Prx1 enhancer element but not from the type II collagen promoter.23 Accordingly, we next focused on the perichondrium, which surrounds the cartilage and is known to synthesize essential factors that regulate endochondral bone growth25, 26, 27 (reviewed in Kronenberg28). To examine gene expression in the perichondrium, we isolated RNA from proximal tibial perichondrium of 2-week-old wild-type, IkkβCol2KO and IkkβPrx1KO mice using laser capture microdissection (LCM). Figure 6a depicts the regions that were subjected to LCM. We confirmed that Ikkβ in the perichondrium was specifically depleted in IkkβPrx1KO but in neither IkkβCol2KO mice nor control wild-type mice (Figure 6b). Then, we examined the expression of genes encoding cytokines that were reported to affect endochondral ossification.17, 29, 30, 31 As shown in Figure 6c, among the cytokines examined, Il-1β and Mcp-5 displayed big differences between IkkβPrx1KO and control mice. Although IL-1β is reported to inhibit the differentiation of chondrocytes,29, 32 it was downregulated in the perichondrium of IkkβPrx1KO mice and thus fails to explain the phenotype. Mcp-5, on the other hand, was upregulated 3.76-fold in IkkβPrx1KO compared with wild-type mice (Figure 6d). Recently, it was proposed that MCP-5 exerts a dual function during development of the joint and growth plate.17 Of note, it was shown that MCP-5 blocks the progression of progenitor mesenchymal cells to chondrocyte hypertrophy and mineralization within the growth plate. Taking these previous findings into consideration, our observation of decreased chondrocyte hypertrophy in IkkβPrx1KO mouse could be explained by an increased production of MCP-5 in the perichondrium. Concordantly, by examining the expression of Ccr2 using RNA isolated from each zone of the growth plate by LCM, we found that Ccr2 can be detected in all zones of the growth plate, but significantly more so in the hypertrophic zone (Figure 6e). Important to note is that CCR2 is the sole receptor for MCP-5.33

Loss of IKKβ in the perichondrium causes aberrant expression of Mcp-5. (a) Before and after LCM of 2-week-old proximal tibial perichondrium (depicted by the green lines). (b) Ikkβ mRNA levels in the perichondrium of control, IkkβF/F;Col2a1-Cre and IkkβF/F;Prx1-Cre mice (n=3 in each group). (c) Cytokine expression in the perichondrium was analyzed with RNA prepared from microdissected samples by RT-PCR. PCR products were run on agarose gel and visualized. (d) qPCR analysis of Mcp-5 transcripts in the perichondrium of control, IkkβF/F;Col2a1-Cre and IkkβF/F;Prx1-Cre mice (n=3 in each group). *P<0.05, two-sided Student’s t-test. Error bars±S.D. (e) Expression of Ccr2 and markers for each zone of the growth plate. The region of each zone after LCM (left panel). Products of RT-PCR for transcripts are indicated (right panel). p, proliferation zone; ph, prehypertrophic zone; h, hypertrophic zone
MCP-5 can inhibit long bone growth ex vivo
These results prompted us to further investigate the role of MCP-5 on the growth of long bone. To this end, we performed tibial organ culture with or without recombinant mouse MCP-5. After 14 or 21 days of culture with recombinant MCP-5, the length of tibiae was significantly shorter than those of control (Figures 7a and b). Histological examination of the bone cultured ex vivo confirmed that the effect on bone length was due to a decreased hypertrophic zone (data not shown), which is consistent with observations of IkkβPrx1KO mouse. To further investigate whether the effect of MCP-5 was exerted through the expression of CCR2 in the hypertrophic zone of the growth plate, we added RS-504393, a specific inhibitor of CCR2, to the ex vivo culture system. As shown in Figure 7c, the addition of RS-504393 cancelled the effect of MCP-5. This result supports the idea that IKKβ loss in the perichondrium overproduces MCP-5, which then remotely affects the growth of long bone by disturbing chondrocyte hypertrophy in the growth plate.

Ex vivo and in vitro experiments demonstrating retardation of bone growth by MCP-5 and the effect of IKKβ on Mcp-5 expression is cell autonomous, respectively. (a) Representative photograph of tibia cultured ex vivo for 14 days. (b) Length of bone after explanted tibiae cultured for 1, 14 and 21 days with or without 10 ng/ml of recombinant mouse Mcp-5 (n=6 in each group). (c) Length of bone after explanted tibiae culture for 14 days with indicated reagents (mouse recombinant MCP-5, 10 ng/ml; RS-504393, 10 μM; n=5 for each group). (d) IkkβF/F-MEFs were infected with control (empty vector), Cre or IκB(SR) harboring retrovirus. At 24 h after infection, cells were treated with or without rhTGF-β1 (2 ng/ml) for 24 h and the mRNA level of Mcp-5 was assessed by RT-qPCR (n=3 for each sample). Data are representative of three independent experiments. *P<0.05, two-sided Student’s t-test. Error bars±S.D.
IKKβ represses Mcp-5 expression in a cell-autonomous manner independently of TβRII and NF-κB
To investigate whether the repression of Mcp-5 expression by IKKβ is direct, and, if so, the underlying mechanism, we performed in vitro studies. We used mouse embryonal fibroblasts (MEFs) for this purpose, because they are of mesenchymal origin. We prepared MEFs from IkkβF/F mice and used retroviral-expressed Cre recombinase to delete Ikkβ exon 3 (Supplementary Figure S1a). We confirmed both the specificity (Supplementary Figure S4a) and the effectiveness (Supplementary Figure S4b) of the deletion. As shown in Figure 7d, deletion of Ikkβ resulted in the upregulation of Mcp-5, which is consistent with our observation in IkkβPrx1KO perichondrium. This result indicates that IKKβ represses Mcp-5 gene expression in a cell-autonomous manner. It was reported that TβRII signaling also regulates Mcp-5 expression during limb development.17 This fact led us to examine the effect of TGF-β1 on the expression of Mcp-5 and its relationship with IKKβ. As shown in Figure 7d, TGF-β1 treatment resulted in the downregulation of Mcp-5 in control (empty vector-treated) cells. Deletion of Ikkβ did not affect this result, indicating that the repressive action of IKKβ on Mcp-5 expression is independent of TGF-β signaling. We also investigated whether the effect of IKKβ is mediated through NF-κB, the major downstream effector of the IKK signalosome. For this purpose, we used IκBα(AA), also known as IκBα super-repressor (SR). In IκBα(SR), the serine 32 and 36 residues of IκBα, the target phosphorylation sites for IKK, are replaced with alanines, resulting in resistance to signal-induced degradation and consequent inhibition of NF-κB activation. When a retroviral vector harboring IκBα(SR) was transduced into IkkβF/F-MEFs, the expression of Mcp-5 was significantly reduced (Figure 7d). This result indicates that NF-κB enhances the expression of Mcp-5 in a signal-dependent manner. Therefore, overall, our results suggest that IKKβ actively represses Mcp-5 expression cell autonomously in a TβRII- and NF-κB-independent manner.
Discussion
Here we report a new role of the IKK complex in bone formation. Contrary to expectations based on the well-known importance of cytokine signaling in the regulation of bone homeostasis,22, 34 we found that IKKβ is dispensable in the osteoblast lineage during normal development and also for osteoprotection during postmenopausal osteoporosis. Chang et al.35 have reported that the inhibition of IKK in the osteoblast lineage by a dominant-negative form of the regulatory subunit IKKγ (also known as NEMO), IKKγ-DN, expressed from the promoter of bone γ-carboxyglutamate protein-2 (Bglap2) or Col1a1 resulted in a substantial increase of trabecular bone mass and bone mineral density without affecting the activities of osteoclasts in young mice.35 In addition, they reported that osteobast lineage-specific expression of IKKγ-DN in mice prevented the osteoporotic bone loss induced by ovariectomy.35 Our different observations cannot be attributed to different genetic backgrounds of the mice, as they too used C57BL/6 mice. Moreover, we used the Col1a1 promoter to specifically drive Cre recombinase in the osteoblast lineage much like how they used it to drive IKKγ-DN, so the inconsistency is not because of differences in the stage of the osteoblast at which IKK was inactivated. The most plausible reason then for the inconsistency is that they overexpressed the IKKγ-DN, whereas we conditionally deleted the gene encoding the catalytic subunit, IKKβ. The C417R mutant of human IKKγ (equivalent to C410R mutant of mouse), which Chang et al.35 used in their study, has been shown to serve as a dominant-negative mutant for cytokine-induced NF-κB activation in human T cells,36 whereas NF-κB activation by lipopolysaccharide is normal when this mutant is expressed in mouse B cells.37 In addition, IKKγ has been shown to control the activation of JNK and p38 MAPK by cooperating with Ubc13 E2 ubiqutin-conjugating enzyme.38 JNK and p38 MAPK are well-known cytokine-responsive MAPKs and are coordinatively activated with NF-κB in response to a wide range of stimuli. Of note, JNK and p38 MAPK are also known to regulate Fra-1, which Chang et al.35 have shown is dysregulated in IKKγ-DN-expressing cells. More recently, it was demonstrated that the expression of IKKγ mutants that allow for IKKβ activation by IL-1β but fail to recruit IκBs results in hyperphosphorylation of alternative IKKβ substrates.39 Therefore, it is highly possible that overexpression of IKKγ-DN might have altered the proper substrate specificity of the IKK complex to have evoked nonphysiological signaling as well as interference with MAPK activation.
Nevertheless, when Ikkβ was conditionally deleted in limb bud mesenchymal cells, the longitudinal bone became shorter due to anomalies with chondrocyte hypertrophy in the growth plate. Therefore, it was surprising when we observed no abnormalities in either cartilage or bone of mice with chondrocyte-specific deletion of Ikkβ. Our results indicate that IKKβ has a role in chondrocyte hypertrophy in a non-cell-autonomous manner. This conclusion begs the question, what could be the cause of the phenotype observed in IkkβPrx1KO mice? Development of the limb skeleton starts with the condensation of undifferentiated mesenchymal cells in the limb bud, which is followed by the aggregated cells proceeding to chondrocyte lineage, whereas cells at the border of condensation form the perichondrium.22, 40 The perichondrium is known to serve as a source of cytokines that regulate bone development.28 Among the cytokines known to affect chondrocytes, we found that MCP-5 is significantly upregulated in the perichondrium of IkkβPrx1KO mice. Mcp-5 is downregulated in embryonic joint-forming interzone cells.17 Moreover, conditional deletion of the gene encoding the TβRII in the limb bud mesenchyme (Tgfbr2Prx1KO) has been shown to result in the upregulation of interzone-MCP-5, and Tgfbr2Prx1KO mice lacked joint development and failed at chondrocyte hypertrophy, suggesting that an unidentified signaling mechanism that represses the expression of Mcp-5 in a TβRII-independent manner counteracts interzone-MCP-5.17 We believe that IKKβ in the perichondrium is this mechanism.
Although our in vitro experiment demonstrated that repression of Mcp-5 by IKKβ is cell autonomous, details of the mechanism are still lacking. In the present study, we showed that the repression of Mcp-5 is NF-κB-independent in unstimulated cells, but that the expression of Mcp-5 can be upregulated in an NF-κB-dependent manner. It could be possible then that the IKK–NF-κB–MCP-5 signaling axis is involved in inflammatory bone diseases such as arthritis, in which cells are chronically exposed to a cytokine stream. Studies combining the use of an Ikkβ conditional knockout (cKO) mutant and disease model mouse may provide an answer to this question.
In conclusion, our results illustrate a unique function of IKKβ in that it remotely controls growth plate development through the surrounding perichondrium (Supplementary Figure S5). This is intriguing because IKKα, the other catalytic subunit of IKK signalosome, has been shown to control skeletal morphogenesis through its action in the epidermis.41
Materials and Methods
Animal studies
All animal experiments were approved by the Kyoto University Animal Care Committee. IkkβF/F mouse was described previously.10 IkkβF/F mice were crossed with Prx1-Cre mice (purchased from Jackson Laboratories, Bar Harbor, ME, USA), Col1a1-Cre mice (obtained from Dr. S Kato, with permission from Dr. G Karsenty, Columbia University Medical Center, New York, NY, USA) and Col2a1-Cre mice (obtained from Dr. N Chano, Shiga University of Medical Science, Shiga, Japan). We genotyped offsprings by PCR using the following primers: Cre-for, 5′-TCCAATTTACTGACCGTACACCAA-3′; Cre-rev, 5′-CCTGATCCTGGCAATTTCGGCTA-3′; IKKβ-flox-for, 5′-GTCATTTCCACAGCCCTGTGA-3′; IKKβ-flox-rev, 5′-CCTTGTCCTATAGAAGCACAAC-3′. The PCR product size was 220 bp (wild-type allele) and 310 bp (floxed allele).10
Histology and histomorphometry
Hind limbs from 2-week-old Ikkβ cKO and IkkβF/F control mice were dissected and used for paraffin and methylmethacrylate (MMA) embedding with and without decalcification, respectively.
For paraffin sections, we fixed tibiae for 20 h in 4% paraformalin, decalcified them in 20% EDTA for 14 days, embedded them in paraffin and obtained 5 μm sections. We stained the sections with safranin-O and used them for immunohistochemistry. For MMA sections, we fixed tibiae for 20 h in 4% paraformalin and embedded them in MMA. Toluidine blue and TRAP staining was performed on MMA sections.
The growth plate of proximal tibia histomorphometry was analyzed using Biorevo (Keyence, Osaka, Japan). Bone histomorphometry was analyzed using a bone morphology image analyzer (Histometry RT CAMERA; System Supply Co., Nagano, Japan).
Skeletal analysis
For alcian blue/alizarin red staining of whole skeletons, 1-day-old Ikkβ cKO and IkkβF/F control mice were stained with alcian blue/alizarin red in glacial acetic acid and 70% ethanol. For three-dimensional bone volume and architecture of the distal femurs, we performed μCT analysis (SMX-100CT-SV3; Shimadzu, Kyoto, Japan) and analyzed the data using the software VGStudio MAX2.0 (Nihon Visual Science Inc., Tokyo, Japan).
Immunohistochemistry
For immunohistochemistry, paraffin sections were rehydrated, and antigens were recovered by treatment with 10 μg/ml of proteinase K for 30 min at 37 °C. Sections were blocked with Protein Blocking (Dako, Tokyo, Japan) and incubated overnight at 4 °C with rabbit anti-type X collagen antibody (LSL, Tokyo, Japan). Alexa Fluor 555 (Invitrogen, Carlsbad, CA, USA) was used for secondary antibody, and sections were counter stained with DAPI.
BrdU and TUNEL assay
Two-week-old mice were injected intraperitoneally with 200 μl (1 ml/100 g of body weight) of BrdU labeling reagent (Zymed Laboratories, South San Francisco, CA, USA) 20 and 4 h before killing. Incorporated BrdU was localized in proximal tibiae by immunohistochemistry using the BrdU Immunohistochemistry System (Chemicon, Darmstadt, Germany) according to the manufacturer’s instructions. Apoptotic cells were detected using the TUNEL-based in situ Cell Death Detection Kit (Roche Diagnostics, Basel, Switzerland). After incubation with 10 μg/ml of proteinase K for 30 min at 37 °C, sections were incubated with the TUNEL reaction mixture for 2 h at 37 °C and mounted with Fluorescence Mounting Medium (Dako).
Microdissection and qPCR
For growth plate chondrocytes and perichondrium, ~40 of 30 μm frozen sections were prepared from tibiae of 2-week-old Ikkβ cKO and Ikkβ floxed mice. Morphologically distinct groups of cells in growth plates were microdissected using PALM MicroBeam (Zeiss, Jena, Germany) and collected in lysis buffer RLT plus reagent (Qiagen, Venlo, Netherlands), and total RNA was extracted with the RNeasy Micro Plus Kit (Qiagen). The quality of RNA was checked with the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Total RNA was reverse-transcribed with SuperScript III (Invitrogen), and one-tenth of the cDNA was amplified by the ABI Prism 7000 sequence detection system (Applied Biosystems) using SYBRgreen (Applied Biosystems, Foster City, CA, USA). Results were normalized to the endogenous expression of Gapdh, and the fold expression was calculated with the 2−ΔΔCT method. Conditions for PCR and qPCR are available upon request.
Residual Ikkβ locus
To evaluate the deleted (IkkβΔ) or residual Ikkβ locus by real-time PCR, a segment of genomic DNA lying at loxP sites or exon 3, respectively, was amplified.19 Schematic representation of the Ikkβ locus and primers are shown in Supplemetary Figure S1a.
Ovariectomy
Twelve-week-old female IKKβ cKO and IkkβF/F control mice were ovariectomized or sham operated. After 8 weeks, all mice were killed and subjected to bone analysis.
Organ culture
Tibiae from 1-day-old mice were harvested and cultured in α-MEM medium (Gibco, Carlsbad, CA, USA) supplemented with 10% FBS for the days indicated in 37 °C/5% CO2. Mouse recombinant Mcp-5 (Cosmo Bio Co., Ltd, Tokyo, Japan) and RS-504393 (Tocris Bioscience, Minneapolis, MN, USA) were added at the indicated final concentrations. Medium was changed every 3days.
Cells and reagents
MEFs were established from IkkβF/F embryos by dissecting E14.5 embryos and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (4.5 g/l glucose) (Nacalai Tesque, Kyoto, Japan) supplemented with 10% FBS. Retroviral vector pMGP-Cre, pMXs-IκBα(AA) and pMXs were transfected to Plat-E cells (generously provided by Dr. T Kitamura) to produce the viral supernatants.
Isolation of primary chondrocytes and osteoblasts
Primary chondrocytes were isolated as described previously.42 Briefly, costal cartilage of 5-day-old mice was digested with collagenase D and cultured in DMEM (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum. Primary osteoblasts were isolated from neonatal mouse calvaria as described previously.43 Calvariae were dissected from neonatal (P2) pups (2nets) and incubated in 4 ml of digestion solution (3.2 mg of collagenase type II (Gibco) per 1 ml of trypsin solution) at 37 °C for 20 min. The supernatant was then transferred to a new tube, and 700 μl of FBS was added to inhibit trypsin activity. The calvaria were washed with 3 ml of DMEM (without FBS), shaken well and added to the supernatant of the tube containing the cell suspension. Cells were collected by centrifugation, resuspended in complete culture medium (DMEM supplemented with 100 μg/ml of ascorbate, 10% FBS (HyClone, Logan, UT, USA) and antibiotic–antimycotic (Gibco)) and seeded into 6-well plates. The procedure was repeated four times.
Statistical analysis
Statistical analyses were performed using Student’s t-test. A P-value of <0.05 was considered statistically significant.
Abbreviations
- IKK:
-
IκB kinase
- NF-κB:
-
nuclear factor-κB
- MCP-5:
-
monocyte chemoattractant protein-5
- TβRII:
-
TGF-β receptor type II
- TGF-β1:
-
transforming growth factor-β1
- BV/TV:
-
bone volume/trabecular volume
- TUNEL:
-
terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling
References
Rothwarf DM, Karin M . The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE 1999; 1999: RE1.
Hacker H, Karin M . Regulation and function of IKK and IKK-related kinases. Sci STKE 2006; 2006: re13.
Karin M, Ben-Neriah Y . Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 2000; 18: 621–663.
Hayden MS, Ghosh S . Shared principles in NF-kappaB signaling. Cell 2008; 132: 344–362.
Chariot A . The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol 2009; 19: 404–413.
Hinz M, Scheidereit C . The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO Rep 2014; 15: 46–61.
Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M et al. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med 1999; 189: 1839–1845.
Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL et al. Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity 1999; 10: 421–429.
Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM . Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science 1999; 284: 321–325.
Li ZW, Omori SA, Labuda T, Karin M, Rickert RC . IKK beta is required for peripheral B cell survival and proliferation. J Immunol 2003; 170: 4630–4637.
Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature 2002; 417: 861–866.
Karsenty G . The complexities of skeletal biology. Nature 2003; 423: 316–318.
Karsenty G, Wagner EF . Reaching a genetic and molecular understanding of skeletal development. Dev Cell 2002; 2: 389–406.
Teitelbaum SL, Ross FP . Genetic regulation of osteoclast development and function. Nat Rev Genet 2003; 4: 638–649.
Boyle WJ, Simonet WS, Lacey DL . Osteoclast differentiation and activation. Nature 2003; 423: 337–342.
Ruocco MG, Maeda S, Park JM, Lawrence T, Hsu LC, Cao Y et al. I{kappa}B kinase (IKK){beta}, but not IKK{alpha}, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med 2005; 201: 1677–1687.
Longobardi L, Li T, Myers TJ, O'Rear L, Ozkan H, Li Y et al. TGF-beta type II receptor/MCP-5 axis: at the crossroad between joint and growth plate development. Dev Cell 2012; 23: 71–81.
Dacquin R, Starbuck M, Schinke T, Karsenty G . Mouse alpha1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn 2002; 224: 245–251.
Egan LJ, Eckmann L, Greten FR, Chae S, Li ZW, Myhre GM et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci U S A 2004; 101: 2452–2457.
Inada M, Matsumoto C, Miyaura C . Animal models for bone and joint disease. Ovariectomized and orchidectomized animals. Clin Calcium 2011; 21: 164–170.
Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ . Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 2002; 33: 77–80.
Kronenberg HM . Developmental regulation of the growth plate. Nature 2003; 423: 332–336.
Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR . Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis 2000; 26: 145–146.
Nishimura I, Chano T, Kita H, Matsusue Y, Okabe H . RB1CC1 protein suppresses type II collagen synthesis in chondrocytes and causes dwarfism. J Biol Chem 2011; 23 286: 43925–43932.
Long F, Linsenmayer TF . Regulation of growth region cartilage proliferation and differentiation by perichondrium. Development 1998; 125: 1067–1073.
Alvarez J, Horton J, Sohn P, Serra R . The perichondrium plays an important role in mediating the effects of TGF-beta1 on endochondral bone formation. Dev Dyn 2001; 221: 311–321.
Colnot C, Lu C, Hu D, Helms JA . Distinguishing the contributions of the perichondrium, cartilage, and vascular endothelium to skeletal development. Dev Biol 2004; 269: 55–69.
Kronenberg HM . The role of the perichondrium in fetal bone development. Ann NY Acad Sci 2007; 1116: 59–64.
Martensson K, Chrysis D, Savendahl L . Interleukin-1beta and TNF-alpha act in synergy to inhibit longitudinal growth in fetal rat metatarsal bones. J Bone Miner Res 2004; 19: 1805–1812.
De Benedetti F, Rucci N, Del Fattore A, Peruzzi B, Paro R, Longo M et al. Impaired skeletal development in interleukin-6-transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum 2006; 54: 3551–3563.
MacRae VE, Farquharson C, Ahmed SF . The restricted potential for recovery of growth plate chondrogenesis and longitudinal bone growth following exposure to pro-inflammatory cytokines. J Endocrinol 2006; 189: 319–328.
Sederquist B, Fernandez-Vojvodich P, Zaman F, Savendahl L . Impact of inflammatory cytokines on longitudinal bone growth. J Mol Endocrinol Apr 2014; 53: T35–T44.
Sarafi MN, Garcia-Zepeda EA, MacLean JA, Charo IF, Luster AD . Murine monocyte chemoattractant protein (MCP)-5: a novel CC chemokine that is a structural and functional homologue of human MCP-1. J Exp Med 1997; 185: 99–109.
Marie PJ . Signaling pathways affecting skeletal health. Curr Osteoporos Rep Sep 10: 190–198.
Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R et al. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med 2009; 15: 682–689.
Yang F, Yamashita J, Tang E, Wang HL, Guan K, Wang CY . The zinc finger mutation C417R of I-kappa B kinase gamma impairs lipopolysaccharide- and TNF-mediated NF-kappa B activation through inhibiting phosphorylation of the I-kappa B kinase beta activation loop. J Immunol 2004; 172: 2446–2452.
Huang TT, Feinberg SL, Suryanarayanan S, Miyamoto S . The zinc finger domain of NEMO is selectively required for NF-kappa B activation by UV radiation and topoisomerase inhibitors. Mol Cell Biol 2002; 22: 5813–5825.
Yamamoto M, Okamoto T, Takeda K, Sato S, Sanjo H, Uematsu S et al. Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat Immunol 2006; 7: 962–970.
Schrofelbauer B, Polley S, Behar M, Ghosh G, Hoffmann A . NEMO ensures signaling specificity of the pleiotropic IKKbeta by directing its kinase activity toward IkappaBalpha. Mol Cell 2012; 47: 111–121.
Akiyama H, Kim JE, Nakashima K, Balmes G, Iwai N, Deng JM et al. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc Natl Acad Sci US A 2005; 102: 14665–14670.
Sil AK, Maeda S, Sano Y, Roop DR, Karin M . IkappaB kinase-alpha acts in the epidermis to control skeletal and craniofacial morphogenesis. Nature 2004; 428: 660–664.
Gosset M, Berenbaum F, Thirion S, Jacques C . Primary culture and phenotyping of murine chondrocytes. Nat Protoc 2008; 3: 1253–1260.
Bakker A, Klein-Nulend J . Osteoblast isolation from murine calvariae and long bones. Methods Mol Med 2003; 80: 19–28.
Acknowledgements
We are indebted to Dr. M Hibi (Nagoya University) for critically reading of the manuscript. We also thank Peter Karagiannis (CiRA, Kyoto University) for help in preparing the manuscript. We thank Dr. G Karsenty for permitting us to use the Col1a1-Cre Tg mouse and Dr. S Kato for providing it, and Dr. T Chano for providing the Col2a1-Cre Tg mouse and Dr. T Kitamura (The University of Tokyo) for providing Plat-E cells. Finally, we thank members of the Toguchida Lab, especially M Ikeya, Y Jin, S Sato, S Tamaki, N Takahara and Y Kobayashi, for their help. This work was supported by Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (21591941 to TK Jr)
Author Contributions
TK Jr, MK and JT conceived the project. KK and TK Jr designed and performed the experiments. KK and TK Jr analyzed the data and wrote the manuscript. MK edited the manuscript. JT supervised the project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
About this article

Cite this article
Kobayashi, K., Toguchida, J., Karin, M. et al. IKKβ in postnatal perichondrium remotely controls endochondral ossification of the growth plate through downregulation of MCP-5. Cell Death Differ 22, 852–861 (2015). https://doi.org/10.1038/cdd.2014.192
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2014.192
This article is cited by
-
CCL12 induces trabecular bone loss by stimulating RANKL production in BMSCs during acute lung injury
Experimental & Molecular Medicine (2023)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}