
Abstract
Aim:
Tanshinol is an important catechol in the antianginal herb Salvia miltiorrhiza roots (Danshen). This study aimed to characterize tanshinol methylation.
Methods:
Metabolites of tanshinol were analyzed by liquid chromatography/mass spectrometry. Metabolism was assessed in vitro with rat and human enzymes. The major metabolites were synthesized for studying their interactions with drug metabolizing enzymes and transporters and their vasodilatory properties. Dose-related tanshinol methylation and its influences on tanshinol pharmacokinetics were also studied in rats.
Results:
Methylation, preferentially in the 3-hydroxyl group, was the major metabolic pathway of tanshinol. In rats, tanshinol also underwent considerable 3-O-sulfation, which appeared to be poor in human liver. These metabolites were mainly eliminated via renal excretion, which involved tubular secretion mainly by organic anion transporter (OAT) 1. The methylated metabolites had no vasodilatory activity. Entacapone-impaired methylation did not considerably increase systemic exposure to tanshinol in rats. The saturation of tanshinol methylation in rat liver could be predicted from the Michaelis constant of tanshinol for catechol-O-methyltransferase (COMT). Tanshinol had low affinity for human COMT and OATs; its methylated metabolites also had low affinity for the transporters. Tanshinol and its major human metabolite (3-O-methyltanshinol) exhibited negligible inhibitory activities against human cytochrome P450 enzymes, organic anion transporting polypeptides 1B1/1B3, multidrug resistance protein 1, multidrug resistance-associated protein 2, and breast cancer resistance protein.
Conclusion:
Tanshinol is mainly metabolized via methylation. Tanshinol and its major human metabolite have low potential for pharmacokinetic interactions with synthetic antianginal agents. This study will help define the risk of hyperhomocysteinemia related to tanshinol methylation.
Similar content being viewed by others
Introduction
In China, Salvia miltiorrhiza roots (Danshen) are extensively used in the treatment of angina pectoris, because the emerging antianginal herbal therapy appears to have low incidence of side effects and may complement the conventional synthetic antianginal therapy to result in a more efficacious strategy1,2,3. Tanshinol (3,4-dihydroxyphenyl lactic acid) is a key compound in Danshen therapies and has been reported to have vasodilatory properties, to protect endothelial cells against homocysteine-induced injury, and to lower methionine-induced hyperhomocysteinemia in rats4,5,6,7,8,9. The other salutary properties of tanshinol include a hypotensive effect, an antithrombotic effect, and myocardial protection against ischemia/reperfusion injury10,11,12. An earlier pharmacokinetic (PK) study indicated that tanshinol, among pharmacologically active Danshen polyphenols, was the only compound to have considerable levels of systemic exposure in human subjects and in laboratory animals after oral administration of cardiotonic pills, a Danshen-containing herbal medicine13. The other Danshen polyphenols were either poorly absorbed from the gastrointestinal tract or eliminated too rapidly; this led to their low plasma concentrations after dosing. In addition to its desirable antianginal and PK properties, tanshinol exhibited very little dose-related toxicity in rats, mice, and dogs14,15.
Because of its catechol structure, tanshinol is expected to undergo catechol-O-methyltransferase (COMT)-mediated methylation. Although several methylated metabolites of tanshinol have been detected in rats after dosing tanshinol16, these metabolites are not fully defined in chemistry and pharmacokinetics. The methylation of xenobiotics with the catechol structure involves COMT-catalyzed transfer of a methyl group from S-adenosylmethionine (SAM) to the catechol hydroxyl group. This transfer produces S-adenosylhomocysteine (SAH), which can be converted to homocysteine by SAH hydrolase17. An abnormally high blood concentration of homocysteine is recognized as a risk factor for cardiovascular diseases and neurodegenerative conditions18,19. This has raised concerns regarding untoward effects of the methylation of xenobiotics on the concentration of homocysteine in the blood20,21. Hyperhomocysteinemia caused by chronic levodopa therapy of Parkinson's disease is a typical example; it is primarily attributed to alteration in the extent of COMT-mediated levodopa methylation22,23,24.
The current study was designed to fully characterize the metabolism of tanshinol, particularly the COMT-mediated methylation, with respect to chemistry, enzyme kinetics, role in tanshinol disposition, and associated interspecies differences. The current study may facilitate a better definition of the safety of Danshen-based therapy and may improve the understanding of the endothelial protection properties of tanshinol. In addition, tanshinol-related herb-drug interaction potential was also assessed with tanshinol and its major human metabolite.
Materials and methods
Chemicals and materials
Tanshinol (sodiated form; purity, ≥98.0%) and norepinephrine bitartrate was obtained from the National Institutes for Food and Drug Control (Beijing, China). Ginsenoside Rg1 and ginsenoside Rb1 were obtained from Tauto Biotech (Shanghai, China). Para-aminohippuric acid, prostaglandin F2α, estrone-3-sulfate, sodium taurocholate, estradiol-17β-D-glucuronide, probenecid, sulfasalazine, methotrexate, rifampin, verapamil hydrochloride, indomethacin, novobiocin, and acetylcholine chloride were obtained from Sigma-Aldrich (St Louis, MO, USA). Entacapone was obtained from Wuhan E-Ternity Technologies (Wuhan, China).
Rat liver cytosol (RLC), rat intestine cytosol (RIC), rat kidney cytosol (RKC), and rat liver microsomes (RLM) were prepared in-house by differential centrifugation. Pooled human liver cytosol (HLC), pooled human liver microsomes (HLM), and cytochrome P450 enzymes inhibitor screening kits for rapid identification of in vitro inhibitors of cDNA-expressed human CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 were obtained from Corning Gentest (Woburn, MA, USA). cDNA-expressed human sulfotransferases SULT1A1, SULT1A3, SULT1B1, SULT1E1, and SULT2A1 were obtained from Sino Biological (Beijing, China). HEK-293 cells (American Type Culture Collection, Manassas, VA, USA) were transfected in-house for transient expression of human organic anion transporter (OAT) 1, OAT2, OAT3, OAT4, organic anion transporting polypeptide (OATP) 1B1, OATP1B3 and rat Oat1, Oat2, and Oat3. Inside-out membrane vesicles [5 mg protein/mL; prepared from insect cells expressing human multidrug resistance-associated protein (MRP) 1, MRP2, MRP3, MRP4, multidrug resistance protein (MDR) 1, and breast cancer resistance protein (BCRP) and rat Mrp1, Mrp2, Mrp4, and Bcrp] were purchased from Genomembrane (Kanazawa, Japan). S-adenosylmethionine (SAM), 3′-phosphoadenosine-5′-phosphosulfate (PAPS), uridine 5′-diphospho-glucuronic acid (UDPGA), and tris-hydroxymethyl-aminomethane (Tris-base) were purchased from Sigma-Aldrich. A reduced nicotinamide adenine dinucleotide phosphate (NADPH)-generating system was comprised of 1.3 mmol/L β-nicotinamide adenine dinucleotide phosphate, 3.3 mmol/L glucose-6-phosphate, 0.5 U/mL glucose-6-phosphate dehydrogenase and 3.3 mmol/L MgCl2.
Experimental animals
All animal care and experimental procedures complied with the Guidance for Ethical Treatment of Laboratory Animals (the Ministry of Science and Technology of China, 2006). The experimental protocols were approved by the Institutional Animal Care and Use Committee at the Shanghai Institute of Materia Medica (Shanghai, China). Male Sprague-Dawley rats (230–270 g; Sino-British SIPPR/BK Laboratory Animal, Shanghai, China) were maintained in a unidirectional airflow room at 20–24°C and a relative humidity of 30%–70% with a 12-h light/dark cycle. The rats were given filtered tap water and commercial rat chow ad libitum and allowed to acclimate to the facilities and environment for three days before use. The rats received in-house femoral-artery-cannulation for blood sampling or bile-duct-cannulation for bile sampling25,26. After surgery, the rats were housed individually and allowed to regain their preoperative body weights before further use. During the bile collection period, a sodium taurocholate solution (pH 7.4) was infused into the duodena of bile-duct-cannulated rats at 1.5 mL/h. After use, the rats were euthanatized with CO2.
Rat PK studies
To detect in vivo metabolites of tanshinol, six femoral-artery-cannulated rats were individually placed in metabolic cages and randomly assigned to two groups of three rats each. The rats received either an intravenous (iv) bolus dose (through the tail veins) or an oral (po) dose (via gavage) of tanshinol at 5 mg/kg. Serial blood samples (∼100 μL; 0, 5, 15, and 30 min and 1, 2, 4, 6, 8, and 24 h after dosing) were collected and heparinized. The blood samples were centrifuged to prepare the plasma fractions. The urine and fecal samples were also collected at -20 °C before and 0–4, 4–8, and 8–24 h after dosing. In addition, six bile-duct-cannulated rats were randomly divided into two groups of three rats each and received either an iv bolus dose (through the tail veins) or a po dose (via gavage) of tanshinol at 5 mg/kg. The bile samples were collected before and 0–4, 4–8, and 8–24 h after dosing. The plasma samples of the same point in time from the same group were pooled and frozen at -70 °C before analysis. The excretory samples were weighed, and the fecal samples were homogenized in 9 volumes of ice-cold saline to produce the fecal homogenate samples. All of the excretory samples of the same type from the same collection period and from the same group were combined and frozen at -70 °C before analysis. The selected doses were those that were commonly used in metabolite detection for drug candidates at the corresponding authors' laboratory in Shanghai (China).
To characterize pharmacokinetics of 3-O-methyltanshinol (M1), 4-O-methyltanshinol (M2), and tanshinol-3-O-sulfate (M7) and to determine influence of methylation on the pharmacokinetics of unchanged tanshinol, femoral-artery-cannulated rats were randomly assigned to four groups of three rats each and received an iv bolus dose of tanshinol at 2 mg/kg (Groups 1 and 3) and a po dose of tanshinol at 10 mg/kg (Groups 2 and 4). The rats in Groups 3 and 4 were pretreated with entacapone (at a po dose of 30 mg/kg) 10 min before dosing tanshinol. Serial blood samples [150 μL; 0, 5, 15, and 30 min and 1, 2, 4, 6 (po tanshinol only), and 8 h (po tanshinol only) after dosing] were collected and heparinized. The blood samples were centrifuged to prepare the plasma fractions. The urine samples were also collected 0–4, 4–8, and 8–24 h after dosing. The plasma and urine samples were stored at -70 °C until analysis. Dose selection was based on the following criteria: (1) the test dose for rats should be as similar to the human daily dose of tanshinol from clinically important Danshen preparations as possible and (2) concentrations of M1 and M7 in rat samples could be accurately measured to generate PK data. According to dose normalization by body surface area27, the iv dose of tanshinol at 2 mg/kg in the rat PK study was roughly equivalent to clinical daily dose of tanshinol from Danshen injection (Z33020177). The po dose of tanshinol at 10 mg/kg was approximately four times clinical daily dose of tanshinol from Danqi tablets (Z43020176).
Because the levodopa-induced hyperhomocysteinemia is mainly attributed to the alteration in extent of levodopa methylation, dosage adjustment was used to generate different methylation extents, which may permit identification of potential hazards for tanshinol in the follow-up study. Rat plasma, liver, and kidney samples were obtained from a recent single ascending dose study of tanshinol (iv; 2–200 mg/kg; equivalent to 1–100 times of the clinical daily dose of tanshinol from Danshen injection)28 and the tanshinol metabolites were measured. In brief, the rats were randomly divided into five groups (12 rats per group), and each group received an iv bolus dose of tanshinol at 2, 5, 15, 50, or 200 mg/kg through the tail veins. The rats were killed under isoflurane anesthesia by bleeding from the abdominal aorta at 5 and 30 min and 1 and 2 h after dosing (three rats per point in time). The blood samples were heparinized and centrifuged to prepare plasma fractions. The kidneys and livers were taken and homogenized.
Detection of tanshinol metabolites
A Waters Synapt G2 high definition time-of-flight mass spectrometer (TOF-MS) (Manchester, UK) was used to detect tanshinol metabolites. This spectrometer was interfaced via a LockSpray source with a Waters Acquity UPLC separation module (Milford, MA, USA). Liquid chromatographic (LC) separation was achieved on a 1.8-μm Waters Acquity UPLC HSS T3 column (100 mm×2.1 mm id; 40 °C; Dublin, Ireland) with mobile phases, delivered at 0.5 mL/min, consisting of A) methanol/water (1:99, v/v; containing 20 mmol/L formic acid) and B) methanol/water (99:1, v/v; containing 20 mmol/L formic acid). A gradient program was used, which consisted of 0–11.0 min from 2% B to 15% B, 11.0–13.0 min at 80% B, and 13.0–18.0 min at 2% B. The mass spectrometer was operated in resolution mode, which gave a resolution power of approximately 20 000. The LockSpray source worked in the negative ion mode. The mass spectrometer was externally calibrated over a range of m/z 50–1000 using a 5 mmol/L sodium formate solution at 20 μL/min, and mass shifts during acquisition were corrected using leucine encephalin (m/z 236.1035 and 554.2615). MSE data acquisition (in centroid mode, m/z 50–1000) was achieved using a trap collision energy of 3 V and a trap collision energy ramp of 15–35 V simultaneously and a scan time of 0.4 s. To support the metabolite detection, an Accelrys metabolite database (version 2012.1; Accelrys, Inc, San Diego, CA, USA) was used to collect prior knowledge of the likely biotransformation pathways of tanshinol. The metabolites of tanshinol were detected in a variety of rat samples and in vitro metabolism incubation samples. Sample preparation was conducted using a methanol-based protein precipitation at a precipitant-to-sample volume ratio of 3:1. After centrifugation, the supernatants were reduced to dryness by centrifugal evaporation under reduced pressure. The residues were reconstituted in 20% methanol for analysis.
Measurement of concentrations of tanshinol metabolites in biomatrices
A Thermo Scientific TSQ Vantage mass spectrometer (San Jose, CA, USA) was used to determine concentrations of 3-O-methyltanshinol (M1), 4-O-methyltanshinol (M2), tanshinol-3-O-sulfate (M7), and tanshinol in rat samples and in a variety of incubation samples. This spectrometer was interfaced via an HESI source with an Agilent Technologies 1290 infinity LC system (Waldbroen, Germany). LC separation for quantification assay was achieved on a Gemini 5-μm C18 column (50 mm×2.0 mm id; Torrance, CA, USA) with mobile phases, delivered at 0.3 mL/min, consisting of A) methanol/water (1:99, v/v; containing 20 mmol/L formic acid) and B) methanol/water (99:1, v/v; containing 20 mmol/L formic acid). A gradient program was used, which consisted of 0–7.0 min from 2% B to 50% B, 7.0–8.5 min from 50% B to 98% B, 8.5–8.6 min from 98% B to 2% B, 8.6–11.0 min at 2% B. The HESI source of the TSQ Vantage mass spectrometer worked in the negative ion mode. The precursor to product ion pairs for tandem mass spectrometry monitoring of M1, M2, M7, and tanshinol were m/z 211→134, 211→134, 277→197, and 197→135, respectively. Sample preparation was conducted using a methanol-based protein precipitation at a precipitant-to-sample volume ratio of 3:1. After centrifugation, the resulting supernatants were applied directly to analysis.
Syntheses of methylated and sulfated metabolites of tanshinol
To characterize the chemical structure of the detected major metabolites of tanshinol, methylated and sulfated products of tanshinol were synthesized and purified. The synthesis of 3-O-methyltanshinol is summarized in Figure 1A. In brief, a benzyl protective group was first introduced into vanillin using potassium carbonate and benzyl bromide. This caused 1 to form. Then, 1 reacted with chloroacetic acid methyl ester under catalysis of benzyltriethylammonium chloride to yield 2. Epoxide cleavage of 2 was induced with boron trifluoride diethyl etherate to produce the ketone 3, which was converted to the alcohol 4 by sodium borohydride. Sodium-hydroxide-based hydrolysis of 4 and the successive adjustment with hydrochloric acid gave the acid 5. The benzyl protective group of 5 was removed using H2 under catalysis of Pd/C to 3-O-methyltanshinol. The synthesis of 4-O-methyltanshinol was accomplished by a procedure that was similar to that for the synthesis of 3-O-methyltanshinol, except that isovanillin served as the starting material instead of vanillin. The synthesis of tanshinol-3-O-sulfate and tanshinol-4-O-sulfate is summarized in Figure 1B. In brief, tanshinol was dissolved in triethylamine/dichloromethane (2:5, v/v) followed by stepwise mixing with pyridine-sulfur trioxide. After stirring for 12 h, the mixture was neutralized with 5% sodium bicarbonate solution and evaporated under reduced pressure. The resulting residue was dissolved in water and neutralized with sodium hydroxide.

Chemical syntheses of 3-O-methyltanshinol (A) and tanshinol-3-O-sulfate/tanshinol-4-O-sulfate (B). Procedure for synthesis of 4-O-methyltanshinol was similar to that for the synthesis of 3-O-methyltanshinol, except that isovanillin served as the starting material instead of vanillin. BF3·Et2O, boron trifluoride diethyl etherate; BnBr, benzyl bromide; ClCH2COOCH3, chloroacetic acid methyl ester; DMF, dimethylformamide; DMSO, dimethyl sulfoxide; NaBH4, sodium borohydride; TEBAc, benzyltriethylammonium chloride; TETN, triethylamine; THF, tetrahydrofuran.
The synthesized compounds were purified by HPLC, and their chemical structures were elucidated by 1H-NMR. The purified synthetic compounds were used to characterize the tanshinol metabolites by LC/TOF-MS with respect to the accurate m/z and retention time.
In vitro metabolism studies of tanshinol
Pilot studies were performed to ensure that comparative capabilities of rat and human cytosolic COMTs were determined for tanshinol under linear metabolite formation conditions with respect to incubation time and protein concentration. Enzyme kinetics of tanshinol methylation was characterized in terms of Michaelis constant (Km), maximum velocity (Vmax), and intrinsic clearance (CLint) by measuring formation of 3-O-methyltanshinol (M1) and 4-O-methyltanshinol (M2). The incubations contained 0.25 mg protein/mL RLC (RIC, RKC, or HLC), 4–1000 μmol/L tanshinol, 50 mmol/L Tris-HCl buffer (pH 7.4), 1 mmol/L dithiothreitol, 2 mmol/L MgCl2, and 1 mmol/L SAM and were maintained for 10 min at 37 °C in duplicate. Before the commencement of the reaction by adding tanshinol, the incubation mixture was preincubated for 3 min at 37 °C. The COMT-mediated methylation of tanshinol (at 1 mmol/L) was further characterized with HLC, RLC, RIC, and RKC by inhibition with entacapone (0–400 nmol/L); half-maximal inhibitory concentrations (IC50) were measured.
Sulfation, glucuronidation, and oxidation of tanshinol and sulfation, glucuronidation, and demethylation of 3-O-methyltanshinol (M1) and 4-O-methyltanshinol (M2) were characterized in vitro by incubation with the human and rat liver subcellular preparations. The sulfation was mediated by SULTs in HLC and RLC fortified with PAPS. The glucuronidation was mediated by UDP-glucuronosyltransferases (UGTs) in HLM and RLM fortified with UDPGA. Cytochrome P450-mediated oxidation of tanshinol and demethylation of M1 and M2 were assessed using NADPH-fortified HLM and RLM. The details of the metabolic incubations have been described previously29,30. After the incubation, the metabolite formation was measured by liquid chromatography/time-of-flight mass spectrometry. The Km, Vmax, and CLint values were determined for tanshinol sulfation under conditions of linear formation of tanshinol-3-O-sulfate (M7). The incubations contained 0.5 mg protein/mL RLC (RIC or HLC), 4–1000 μmol/L tanshinol, 50 mmol/L Tris-HCl buffer (pH 7.4), 8 mmol/L dithiothreitol, 5 mmol/L MgCl2, and 1 mmol/L PAPS and were maintained for 60 min at 37 °C in duplicate. cDNA-expressed human SULT1A1, SULT1A3, SULT1B1, SULT1E1, and SULT2A1 were assessed with respect to formation of M7 by incubation for 120 min; the tanshinol concentration was 500 μmol/L. The Km, Vmax, and CLint values were also determined for M1 sulfation and M2 sulfation under conditions of linear formation of 3-O-methyltanshinol sulfate (M3) and 4-O-methyltanshinol sulfate (M4), respectively. The incubations contained 0.5 mg protein/mL RLC (RIC or HLC), 4–5000 μmol/L M1 (or M2), 50 mmol/L Tris-HCl buffer (pH 7.4), 8 mmol/L dithiothreitol, 5 mmol/L MgCl2, and 1 mmol/L PAPS and were maintained for 60 min at 37 °C in duplicate experiments.
Cellular and vesicular transport assessment of tanshinol metabolites
Cellular uptake methods for assessment of 3-O-methyltanshinol (M1), 4-O-methyltanshinol (M2), and tanshinol-3-O-sulfate (M7) are similar to those for assessment of tanshinol28. In brief, sequence-verified rat Oat1, Oat2, and Oat3 and human OAT1, OAT2, OAT3, and OAT4/pcDNA3.1(+) constructs and empty vector were transiently transfected into HEK-293 cells with the lipofectamine 2000 transfection reagent according to the manufacturer's protocol. Uptake and inhibition studies were carried out in 24-well poly-D-lysine-coated plates with the cells 48 h after transfection. Before use, the transfected cells were functionally characterized with para-aminohippuric acid (a substrate for Oat1 and OAT1), prostaglandin F2α (a substrate for Oat2 and OAT2), estrone-3-sulfate (a substrate for Oat3, OAT3, and OAT4), and probenecid (an inhibitor for the Oat and OAT transporters). Differential cellular uptake between the transfected cells (TC) and the mock cells (MC) was defined as net transport ratio (TransportTC/TransportMC ratio), and a net transport ratio greater than three suggested a positive result. Kinetics of transporter-mediated cellular uptake of M1, M2, and M7 were assessed with respect to Km, Vmax, and CLint. Initially, the incubation time was optimized to ensure that the assessment was performed under linear cellular uptake conditions. The uptakes of M1, M2, and M7 (39–5000 μmol/L) were determined in transfected cells or mock cells over an optimal 5-min period. The difference in amount of compound transported into the transfected cells and the mock cells served as the transporter-dependent uptake for determination of transport kinetics. Membrane vesicles expressing one of the rat Mrp1, Mrp2, Mrp4, and Bcrp and human MRP1, MRP2, MRP3, MRP4, and BCRP were used for assessment of M1, M2, and M7 by a rapid filtration method that was similar to the method used for tanshinol28. A net transport ratio (TransportATP/TransportAMP ratio) of greater than two was considered positive for ATP-dependent transport.
Assessment of potential of tanshinol and its major human metabolite to inhibit human cytochrome P450 enzymes and human OATP1B1/3, MDR1, MRP2, and BCRP transporters
Inhibitory effects of tanshinol and 3-O-methyltanshinol (M1) on 3-cyano-7-ethoxycoumarin O-deethylation (CYP1A2 and CYP2C19), coumarin 7-hydroxylation (CYP2A6), 7-ethoxy-4- trifluoromethylcoumarin O-deethylation (CYP2B6), dibenzylfluorescein O-debenzylation (CYP2C8), 7-methoxy-4-trifluoromethylcoumarin O-demethylation (CYP2C9), 3-[2-(N,N-diethyl-N-methylamino)ethyl]-7-methoxy-4-methylcoumarin O-demethylation (CYP2D6), and 7-benzyloxy-trifluoromethylcoumarin O-debenzylation (CYP3A4) activities were evaluated using cDNA-expressed human P450 enzymes. As positive controls, the IC50 values of furafylline against CYP1A2; tranylcypromine against CYP2A6, CYP2B6, and CYP2C19; quercetin against CYP2C8; sulfaphenazole against CYP2C9; quinidine against CYP2D6; and ketoconazole against CYP3A4 were 2.9, 0.07, 5.9, 7.8, 9.5, 0.2, 0.005, and 0.007 μmol/L, respectively. The initial concentrations of the test compounds were 100 μmol/L. The IC50 value was not further determined when demonstrating <50% inhibition.
The inhibitory effects of tanshinol and 3-O-methyltanshinol (M1) on the activities of human OATP1B1 and OATP1B3 (mediating transport of estradiol-17β-D-glucuronide) were also assessed with transiently transfected HEK-293 cells. The inhibitory effects on the activities of human MDR1 (mediating transport of ginsenoside Rg1), human MRP2 (mediating transport of sulfasalazine), and human BCRP (mediating transport of methotrexate) were also assessed with corresponding membrane vesicles. The detailed methods have been described previously31. The inhibitors of positive control included rifampin (OATP1B1), ginsenoside Rb1 (OATP1B3), verapamil (MDR1), indomethacin (MRP2), and novobiocin (BCRP). The initial concentrations of the test compounds were 100 μmol/L. The IC50 value was not further determined when the compound demonstrated <50% inhibition.
Assessments of vasodilatory activities of tanshinol metabolites
After intraperitoneal pentobarbital (50 mg/kg) anesthesia, proximal descending thoracic aortae were isolated from rats (260–300 g), cleaned of adherent tissue, and cut into rings (3–4 mm). The aortic rings were endothelium-intact and suspended in ice-cold Krebs-Henseleit buffer (118 mmol/L NaCl, 4.7 mmol/L KCl, 1.2 mmol/L MgSO4, 1.2 mmol/L KH2PO4, 1.5 mmol/L CaCl2, 25 mmol/L NaHCO3, 10 mmol/L D-glucose, and 0.05 mmol/L EDTA; pre-gassed with 95% O2/5% CO2). Variations in isometric tension of the aortic rings were recorded under 2-g optimal resting tension using a Radnoti 159901A isometric force transducer (Radnoti LLC, Monrovia, CA, USA) and a PowerLab 8/30 (AD Instruments, Sydney, Australia). The aortic rings were placed between two stainless-steel stirrups that were immersed in Krebs-Henseleit buffer (37 °C; contained in 20-mL organ chambers). The buffer was gassed continuously with 95% O2/5% CO2. After equilibrium for 60 min, the aortic rings were checked three times for a reproducible contractile response to 60 mmol/L KCl (each followed by wash in Krebs-Henseleit buffer). The integrity of the endothelium in each aortic ring was determined by an adequate relaxation response (>70%) to 10 μmol/L acetylcholine on the 1-μmol/L norepinephrine-precontracted tone. The qualified aortic rings were retreated with norepinephrine (1 μmol/L) to establish a stable contractile tone. Relaxation responses of 3-O-methyltanshinol (M1), 4-O-methyltanshinol (M2), and tanshinol-3-O-sulfate (M7) were measured by cumulative addition of these compounds individually into the organ chambers at 10-min intervals and compared with those of tanshinol.
Data processing
PK parameters were determined using non-compartmental analysis with a Thermo Kinetica software package (version 5.0; Philadelphia, PA, USA). The details have been described previously25. The metabolic clearance via methylation (CLm-M) was calculated by dividing the sum of Cum.Ae-U values of 3-O-methyltanshinol (M1) and 4-O-methyltanshinol (M2) by the AUC0-∞ of unchanged tanshinol. The metabolic clearance via sulfation (CLm-S) was calculated by dividing the Cum.Ae-U of tanshinol-3-O-sulfate (M7) by the AUC0-∞ of unchanged tanshinol. GraFit software (version 5; Surrey, UK) was used to determine Km, Vmax, and IC50 values30. All data are expressed as the mean±standard deviation (SD). Statistical analysis was performed with IBM SPSS Statistics Software (version 19.0; Chicago, IL, USA). A value of P<0.05 was considered to be the minimum level of statistical significance.
Results
Metabolism of tanshinol in rats and chemical structures of the major metabolites
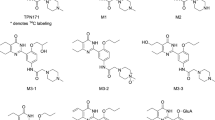
Guided by the computational prediction of tanshinol metabolism, a total of eight possible metabolites of tanshinol, M1–M8, were detected in rat plasma, urine, and bile samples after a single iv or po dose of tanshinol at 5 mg/kg, but only M1–M3, and M7 were detected in the fecal samples (Table 1). Neither these metabolite candidates nor unchanged tanshinol was detected in rat samples before dosing. LC/TOF-MSE-based detection abundances of the metabolite candidates and unchanged tanshinol in plasma decreased rapidly after iv dosing. These metabolite candidates of tanshinol were detected during the first 4 h after dosing. After po dosing, the detection abundances of the metabolite candidates and unchanged tanshinol in plasma increased and then decreased, peaking at 0.5–1 h; these compounds remained detectable for 8 h. These PK behaviors suggested that the detected metabolite candidates were the metabolites of tanshinol in rats. The accurate molecular masses of M1 and M2 were consistent with those of the predicted methylated metabolites of tanshinol, M3 and M4 with the methylated sulfates of tanshinol, M5 and M6 with the methylated glucuronides of tanshinol, M7 with the sulfate of tanshinol, and M8 with the glucuronide of tanshinol. Neither an oxidized metabolite nor a decarboxylated metabolite of tanshinol was detected in any rat sample after dosing. The detected metabolites of tanshinol were predominantly recovered in the rat urine samples 0–8 h after dosing but were poorly detected in the bile samples. This suggested that renal excretion was their major route of elimination. Among the detected metabolites, M1 and M7 were two major circulating metabolites of tanshinol; they were also major urinary metabolites of tanshinol. These results suggested that methylation and sulfation were the major metabolic pathways of tanshinol in rats. The iv and po dosed tanshinols exhibited similar metabolite profiles, except for M7 showing an AUCiv (M7)/AUCiv (tanshinol) ratio that was smaller than the AUCpo (M7)/AUCpo (tanshinol) ratio.
As shown in Table 2, incubation of tanshinol with SAM-fortified RLC resulted in formation of M1 and M2; incubation with PAPS-fortified RLC resulted in formation of M7. These in vitro metabolism data further confirmed the metabolic nature of these tanshinol metabolites detected in the rats. To elucidate the chemical structures of these tanshinol metabolites, 3-O-methyltanshinol, 4-O-methyltanshinol, tanshinol-3-O-sulfate, and tanshinol-4-O-sulfate were synthesized (Figure 1); the purified synthetic products were characterized by 1H-NMR and LC/TOF-MSE (Table 3). By comparing the LC/TOF-MSE data of the synthetic products with those of the tanshinol metabolites that were detected in the rats, M1, M2, and M7 were characterized as 3-O-methyltanshinol, 4-O-methyltanshinol, and tanshinol-3-O-sulfate, respectively. The other tanshinol metabolites M3–M6 and M8 were also characterized using in vitro metabolic reactions that were mediated by rat cytosolic or microsomal enzymes, and the results are shown in Table 2. Incubation of M1 and M2 with PAPS-fortified RLC resulted in formation of M3 and M4, respectively; this indicated that M3 and M4 were 3-O-methyltanshinol sulfate and 4-O-methyltanshinol sulfate, respectively. Incubation of M1, M2, and tanshinol with UDPGA-fortified RLM resulted in formation of M5, M6, and M8, respectively, indicating that M5, M6, and M8 were 3-O-methyltanshinol glucuronide, 4-O-methyltanshinol glucuronide, and tanshinol glucuronide, respectively. P450-mediated oxidation of tanshinol and demethylation of M1 and M2 were not detected after incubation with NADPH-fortified RLM. Figure 2 depicts the proposed metabolic pathways of tanshinol in rats. Supplementary Information Figure S1 shows the typical LC-MS chromatograms and MSE spectra of the tanshinol metabolites.

Proposed metabolic pathways of tanshinol in rats.
Enzyme kinetics of methylation and other metabolisms of tanshinol
Methylation of tanshinol by incubation with SAM-fortified RLC was saturable with the Km, Vmax, and CLint values for the formation of 3-O-methyltanshinol (M1) and 4-O-methyltanshinol (M2) are shown in Table 4. The rat hepatic COMT-mediated methylation resulted in a predominant formation of M1 over M2 with an M1-to-M2 CLint ratio of 9.9. Rat renal and intestinal COMTs also exhibited catalytic activities towards tanshinol methylation, but their activities were lower than that of their hepatic counterpart. M1-to-M2 CLint ratios were 13.0 for RKC and 11.6 for RIC. The CLint values were ranked as follows: RLC (100% for M1 and 100% for M2)>RKC (38% and 29%)>RIC (10% and 9%). The differences in CLint were primarily associated with Vmax rather than Km. Rat hepatic, renal, and intestinal COMT-mediated methylation of tanshinol could be inhibited by entacapone with IC50 values of 23.7, 25.7, and 24.6 nmol/L, respectively. Both RLC and RIC exhibited significant catalytic activity towards tanshinol sulfation, but RKC did not have any such catalytic activity; the Km, Vmax, and CLint values are shown in Table 4. The CLint of tanshinol sulfation by RIC was 18% of that of RLC. COMT and SULT have been reported to have concerted actions in metabolism of catechol drugs32,33. Sulfation of 4-O-methyltanshinol and that of 3-O-methyltanshinol exhibited significantly different metabolic capabilities (CLint), which were mainly attributed to the difference in Vmax, rather than Km (Table 4). The CLint values for 4-O-methyltanshinol sulfation and 3-O-methyltanshinol sulfation were 2.3 times and 3% of tanshinol sulfation, respectively. Because M2 was a minor metabolite of tanshinol in rats and because the sulfation of M1 was quite slow, neither 3-O-methyltanshinol sulfate (M3) nor 4-O-methyltanshinol sulfate (M4) was considerably detected in rats after dosing tanshinol (Table 1).
To facilitate extrapolation of the rat metabolic data of tanshinol into human situation, the metabolism of tanshinol was also assessed in vitro with human enzymes. Methylation of tanshinol by incubation with SAM-fortified HLC also resulted in a predominant formation of M1 over M2; the Km, Vmax, and CLint values are shown in Table 4. This process could be inhibited by entacapone with an IC50 of 8.5 nmol/L. Unlike RLC, HLC (fortified with PAPS) exhibited low sulfation activity towards tanshinol to form M7. The observed interspecies difference in CLint was associated with lower affinity (Km) and a lower Vmax for HLC compared with those for RLC (Table 4). Because there is considerable variation in expression patterns of SULTs in different human tissues34, a variety of cDNA-expressed human SULT enzymes were tested. Three human SULTs exhibited catalytic activity towards tanshinol sulfation into M7; these SULTs were ranked as follows: SULT1A3 (100%; highly expressed in the human intestine but not in the liver)>SULT1A1 (30%)>SULT1B1 (18%). Sulfation of M1 into M3 and that of M2 into M4 were poorly mediated by human hepatic SULTs (Table 4).
Renal excretion, herb-drug interaction potential and vasodilatory activities of tanshinol metabolites
Overall mean recovery of iv dosed tanshinol in rats was 88.0% in urine [including 59.6% as unchanged tanshinol, 17.8% as 3-O-methyltanshinol (M1), 0.9% as 4-O-methyltanshinol (M2), and 9.7% as tanshinol-3-O-sulfate (M7)] and 0.2% in bile (including 0.01% as unchanged tanshinol, 0.14% as M1, and negligible M2 and M7) (Table 5). The CLR values of M1, M2, and M7 (3.6–4.1 L·h−1·kg−1) were slightly greater than that of unchanged tanshinol (2.0 L·h−1·kg−1). The fractions of plasma protein-unbound M1, M2, M7, and tanshinol (fu) were 83%, 72%, 32%, and 98%, respectively, which were measured by a rapid ultrafiltration method35. Like tanshinol, the renal excretion of M1, M2, and M7 in rats mainly involved tubular secretion, rather than glomerular filtration. This was indicated by their CLR/(GFR×fu) ratios, which were 19, 22, and 42, respectively. Because of the involvement of active tubular secretion, the cellular and vesicular uptakes of M1, M2, and M7 by renal transporters were evaluated. Similar to tanshinol, M1 and M2 were transported by rat Oat1, and the Km values are shown in Table 4. Rat Oat3 also exhibited transport activities towards M1 and M2, but with lower affinity than that of Oat 1; the Km values of M1 and M2 for rat Oat2 were even greater. Both Oat1 and Oat3 mediated cellular uptake of M7; Oat1 exhibited greater Km and Vmax than Oat3 (Table 4). Oat2 did not exhibit any considerable transport activity towards M7. Similar to tanshinol, rat Mrp2, Mrp4, and Bcrp did not mediate any significant vesicular uptake of M1, M2, and M7. In addition to the rat renal transporters, human renal transporters were also evaluated with respect to their cellular and vesicular uptake of M1, M2, and M7; the transporter kinetics data are shown in Table 4. These tanshinol metabolites were considerably transported by human OAT1; human OAT2 and OAT4 also exhibited cellular uptake activities but with lower affinity; human OAT3 exhibited cellular uptake activities only towards M1 and M2. Human MRP1, MRP2, MRP3, MRP4, and BCRP did not mediate any significant transport of M1, M2, and M7.
Recently, efforts have been directed at evaluating the potential inhibitory effects of both parent drugs and their metabolites on the common drug metabolizing enzymes and transporters and the contribution of metabolites to in vivo drug-drug interactions36,37,38,39,40. The use of Danshen therapy is prevalent among patients with angina pectoris who are also taking synthetic antianginal agents, the disposition of which often involves cytochrome P450 enzymes and many hepatic transporters41,42,43. Accordingly, tanshinol and its major circulating metabolite 3-O-methyltanshinol (M1) were assessed with respect to their inhibitory potential against human cytochrome P450 enzymes (including CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) and human transporters (OATP1B1, OATP1B3, BCRP, MRP2, and MDR1). As a result, tanshinol and M1 (100 μmol/L for each) did not exhibit any significant in vitro inhibitory activity towards these drug-metabolizing enzymes and transporters, except for CYP2C8, CYP2C9, and CYP2C19. Tanshinol was found to have weak in vitro inhibitory activities against CYP2C8, CYP2C9, and CYP2C19 with IC50 values of 34, 99, and 67 μmol/L, respectively.
Methylation and sulfation are often pathways for inactivation of xenobiotics44,45. Unlike tanshinol, 3-O-methyltanshinol (M1) and 4-O-methyltanshinol (M2) were found to have no vasodilatory property on rat thoracic arteries that were precontracted by norepinephrine (Figure 3). Tanshinol-3-O-sulfate (M7) exhibited only a weak vasodilatory activity. Although more pharmacological assessments are warranted for these tanshinol metabolites, the overall antianginal effect of tanshinol might be attributed primarily to unchanged tanshinol.

Vasodilatory effects of 3-O-methyltanshinol (M1), 4-O-methyltanshinol (M2), tanshinol-3-O-sulfate (M7), and unchanged tanshinol on norepinephrine-induced contraction of rat thoracic arteries. Each measure represents the mean value of four or five technical replicates.
Influence of methylation on systemic exposure to tanshinol and dose-related saturation of tanshinol methylation in rats
Figure 4 shows the mean plasma concentrations of 3-O-methyl-

Plasma concentrations of 3-O-methyltanshinol (M1; green curves/open circles), 4-O-methyltanshinol (M2; green curves/solid circles), tanshinol-3-O-sulfate (M7; brown curves), and unchanged tanshinol (blue curves) over time after an iv (2 mg/kg) and a po dose of tanshinol (10 mg/kg) in rats not treated with (A and B) or treated with oral entacapone at 30 mg/kg (C and D). Panels E and F show comparative plasma AUC0–∞ of M1 (green open bars), M2 (green solid bars), M7 (brown bars), and unchanged tanshinol (blue bars) in rats not treated with entacapone and rats treated with entacapone. BLQ, blow the lower limit of quantification. The data represent means and standard deviations (n=3). bP<0.05 compared with the rat group that was not treated with entacapone.
tanshinol (M1), 4-O-methyltanshinol (M2), tanshinol-3-O-sulfate (M7), and unchanged tanshinol over time after an iv bolus dose (2 mg/kg) or a po dose (10 mg/kg) of tanshinol in rats. Plasma PK parameters and associated urinary and biliary excretion data of the tanshinol metabolites are summarized in Table 5. The plasma concentrations of M1, M2, and M7 changed concurrently with those of tanshinol after dosing. These metabolites were eliminated rapidly with mean t1/2 values of 0.17–0.18 h after iv dosing. Mean plasma AUC0-∞ values of M1, M2, and M7 were 14.7%, 0.7%, and 9.2% of that of unchanged tanshinol, respectively, after iv dosing and 29.0%, 1.0%, and 37.6%, respectively, after po dosing, suggesting that the intestine was probably a considerable site of tanshinol metabolism, particularly for sulfation. After iv dosing, mean apparent metabolic clearance of tanshinol via methylation (CLm–M) and that via sulfation (CLm–S) accounted for 18.8% and 9.9% of the plasma total clearance (CLtot, p) of tanshinol, respectively.
Drug methylation can be substantially impaired in vivo by COMT inhibitors46. To determine the influence of COMT-mediated methylation on systemic exposure to unchanged tanshinol, rats were treated with oral entacapone (30 mg/kg), a potent COMT-inhibitor, to impair the methylation of tanshinol. As shown in Figure 4 and Table 5, entacapone treatment caused plasma 3-O-methyltanshinol (M1) and 4-O-methyltanshinol (M2) to be very little or not to be detected after an iv bolus dose (2 mg/kg) or a po dose (10 mg/kg) of tanshinol. Consistent with these systemic exposure data, the mean fraction of dose excreted into urine (fe–U) as M1 was significantly lower in the rats treated with entacapone than in the same rats not treated with entacapone, demonstrating 0.4% versus 17.8% for iv dosing (P=0.001) and 0.7% versus 7.9% for po dosing (P=0.03). Urinary M2 was not detected after entacapone treatment, except for the first urine sample of 0–4 h after iv dosing. The metabolic clearance of tanshinol via methylation (CLm–M) decreased from 0.63 to 0.01 L·h−1·kg−1 after entacapone treatment (P=0.00004). Although entacapone impaired the methylation of tanshinol in rats, it did not significantly enhance rat systemic exposure to tanshinol. This was indicated by mean AUC0–∞ values of tanshinol in rats treated with entacapone, which were only 1.34-fold (for iv dosing; P=0.01) and 1.43-fold (for po dosing; P=0.1) of the respective values of tanshinol in the same rats not treated with entacapone. The limited influence of entacapone treatment on the systemic exposure to unchanged tanshinol appeared to result from considerable compensation by the SULT-mediated sulfation to form tanshinol-3-O-sulfate (M7) and the renal excretion of unchanged tanshinol. This was indicated by mean plasma AUC0-∞ values of M7, which were increased 4.0-fold (for iv dosing; P=0.01) and 2.1-fold (for po dosing; P=0.001) by entacapone treatment. Consistently, the urinary fe-U values of M7 in rats treated with entacapone were 3.0-fold (for iv dosing; P=0.005) and 1.8-fold (for po dosing; P=0.01) greater than those of the same rats not treated with entacapone. Entacapone treatment also resulted in increases of fe-U of tanshinol from 59.6% to 67.7% (for iv dosing; P=0.1) and from 12.8% to 17.7% (for po dosing; P=0.02).
Figure 5 shows dose-dependent levels of systemic, liver, and kidney exposure to 3-O-methyltanshinol (M1), 4-O-methyltanshinol (M2), tanshinol-3-O-sulfate (M7), and unchanged tanshinol in rats. Over the iv dose range 2–200 mg/kg, metabolite-to-tanshinol ratios in plasma C5 min were 0.05–0.1 for M1 and M7, and such ratios in plasma AUC0-∞ were 0.1–0.2. Unlike unchanged tanshinol, M1 and M7 exhibited higher levels of liver exposure than the respective levels of systemic exposure. Over the iv dose range 2–50 mg/kg, metabolite-to-tanshinol ratios in liver C5 min were 5.6–10.5 and 3.3–6.6 for M1 and M7, respectively; such ratios in liver AUC0-∞ were 4.3–7.8 and 2.3–4.4, respectively. However, at the iv dose 200 mg/kg, the liver concentration of unchanged tanshinol increased steeply to levels that were significantly higher than the liver concentrations of M1 and M7, and the metabolite-to-tanshinol ratios in C5 min and in AUC0-∞ were <1.0 for M1 and M7. This result indicated that tanshinol methylation and sulfation in the rat liver became saturated as the dose increased to 200 mg/kg. Like unchanged tanshinol, M1 and M7 exhibited higher levels of kidney exposure than the respective levels of systemic exposure. Metabolite-to-tanshinol ratios in kidney C5 min and AUC0-∞ for M1 decreased from 0.60 to 0.08 and from 0.98 to 0.17, respectively, as the dose increased from 2 to 200 mg/kg. Such ratios in kidney C5 min and AUC0–∞ for M7 decreased from 0.21 to 0.05 and from 0.30 to 0.08, respectively, indicating that the levels of kidney exposure to M7 were lower than the respective levels of kidney exposure to M1. The plasma, liver, kidney C5 min and AUC0-∞ of M2 were 1%–7% of those of M1. The dose-dependent levels of systemic, liver, and kidney exposure to M1, M2, M7, and unchanged tanshinol are summarized in Table 6.

Levels of rat systemic (A and B), liver (C and D), and kidney exposure (E and F) to 3-O-methyltanshinol (M1; green curves/open circles), 4-O-methyltanshinol (M2; green curves/solid circles), tanshinol-3-O-sulfate (M7; brown curves), and tanshinol (blue curves) over iv bolus dose of tanshinol (2–200 mg/kg). The data represent means and standard deviations from an experiment, in which each rat dose group had four sampling times (each in triplicate).
Discussion
Tanshinol is a key compound in antianginal Danshen therapy because it had desirable antianginal and PK properties4,5,6,7,8,9,10,11,12,13. The current study and a recent study by Jia et al28 revealed how tanshinol was eliminated at the molecular level; the major elimination processes were renal excretion and methylation. The renal excretion of tanshinol involved active tubular secretion and glomerular filtration with CLR/(GFR×fu) values of 5.0 for human subjects and 5.6 for rats. The tubular secretion of tanshinol consisted of the basolateral uptake from blood, primarily by human OAT1 and rat Oat1, and the subsequent luminal efflux into urine, mainly by passive diffusion. Human basolateral OAT2/OAT3 and rat basolateral Oat3 also played considerable roles in the tubular secretion when tanshinol concentration in blood was high. Because of its catechol structure, tanshinol was also eliminated by COMT-mediated methylation. Zhang et al reported four methylated metabolites that were detected in rat plasma and urine after dosing tanshinol16. However, in the current study, only 3-O-methyltanshinol (M1) and 4-O-methyltanshinol (M2) were detected. To confirm whether any other methylated metabolites of tanshinol were formed in the rats, 8-O-methyltanshinol and tanshinol methyl ester were synthesized. However, these two synthesized compounds were not detected in any rat sample after the iv or po administration of tanshinol (data not shown). Also in human subjects, only two methylated metabolites of tanshinol were detected in plasma and urine after iv dosing DanHong injection, a Danshen-containing herbal injection47. Both rat and human COMT favored the 3-O-methylation of tanshinol over the 4-O-methylation. The methylated metabolites of tanshinol were eliminated via renal excretion, mainly involving OAT- or Oat-mediated tubular secretion. The preceding renal excretion and methylation work in concert but compete with each other; their roles in the elimination of tanshinol depend on the sizes of CLR and CLm–M. Figure 6 depicts the mechanistic elimination of tanshinol.

A metabolism/transport map of clearance of tanshinol (TSL). It shows the chemical fate, appearance and disappearance of tanshinol and its metabolites 3-O-methyltanshinol (M1), 4-O-methyltanshinol (M2), and tanshinol-3-O-sulfate (M7) from the principal compartments, and associated drug metabolizing enzymes and drug transporters. COMT, catechol-O-methyltransferase; SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; SULT, sulfotransferase; PAPS, 3′-phosphoadenosine-5′-phosphosulphate; PAP, 3′-phosphoadenosine-5′-phosphate; OAT/Oat, organic anion transporter; tHcy, total homocysteine.
Pharmacotherapy for angina pectoris normally involves alleviation of symptoms of myocardial ischemia plus treatment of cardiac risk factors48,49. In addition to having the desirable pharmacological properties, an ideal antianginal agent should have minimal toxicity and a low propensity to cause drug interactions. These properties are underscored in angina patients, of whom a significant number would have long-term and multiple medications, particularly for effective reduction of progression of the ischemic heart disease. Full hazard identification and risk assessment of tanshinol are essential to support long-term use of the Danshen-based antianginal therapy. Abnormal blood homocysteine concentration, due to an imbalance in the homocysteine generation-elimination relationship, is recognized as a risk factor for cardiovascular diseases and neurodegenerative conditions18,19. The only known mechanism of homocysteine production involves SAM-dependent compound methylation that produces SAH. The resulting SAH can be converted into homocysteine by SAH hydrolase. Drug methylation may affect the blood concentration of homocysteine50; a typical example is hyperhomocysteinemia induced by chronic levodopa therapy of Parkinson's disease22,23,24. The levodopa therapy normally involves coadministration of carbidopa, a peripheral dopa decarboxylase inhibitor, to increase brain dopamine levels. However, the inhibition of decarboxylation leads to a shift of levodopa metabolism towards methylation. This shift can enhance SAH generation and induce hyperhomocysteinemia. Like levodopa, tanshinol also has a catechol structure. The full characterization of tanshinol methylation will help to better define the safety of Danshen-based therapy.
The preceding levodopa story suggests that the hyperhomocysteinemia is related to alteration in the extent of levodopa methylation. The extent or magnitude of tanshinol methylation in the body depended on factors such as metabolic capacity of COMT, competition of methylation with other concurrent elimination pathways, and tanshinol dose. Although rat and human soluble COMTs share 80% similarity in amino acid sequences with the active site highly conserved51, rat hepatic COMT exhibited lower affinity for tanshinol (Km, 549 μmol/L) than human hepatic COMT (51 μmol/L). However, the intrinsic metabolic capacity (CLint) for tanshinol methylation in rats was not very different from that in humans (Table 4). This was because the rat COMT had significantly greater Vmax values (3598 and 378 pmol/mg protein/min for the formation of M1 and M2, respectively) than human COMT (138 and 26 pmol/mg protein/min, respectively). In addition, competition with concurrent renal excretion could counterbalance the influence of interspecies difference in affinity of tanshinol for rat and human COMTs on the extent of tanshinol methylation. After iv administration of the tanshinol-containing DanHong injection, the CLm–M of tanshinol accounted for only 18.1% and 13.7% of its CLtot, p in rats and human subjects, respectively; the CLR of tanshinol accounted for 54.3% and 63.6%, respectively47. Accordingly, rats could be used to investigate hyperhomocysteinemia risk related to tanshinol methylation. Rats have been used to study the management of levodopa-induced hyperhomocysteinemia52,53. The results of the single ascending dose study of iv tanshinol in rats (Figure 5 and Table 6) suggested that the saturation of tanshinol methylation in the liver could be predicted from the Km for COMT in RLC. Although both the liver concentrations of unchanged tanshinol and M1 clearly reflected the saturation, only the plasma concentration of M1, rather than the plasma concentration of tanshinol, could reflect the saturation. This is because the influence of saturation of tanshinol methylation could be compensated by tanshinol's renal excretion. Tanshinol methylation, rather than sulfation, also took place in the rat kidney. Saturation of tanshinol methylation in the rat kidneys appeared to occur after iv dosing at a low level, because of the action of rat renal Oat1/3. The kidney C5 min of tanshinol after iv dosing tanshinol at 15 mg/kg exceeded the Km for COMT in RKC. A similar scenario is expected to take place in humans. To define the risk of hyperhomocysteinemia related to tanshinol methylation, it would be helpful to identify PK markers to reflect the methylation extent or magnitude. Herein, the results of entacapone-impaired and high-dose-saturated methylation of tanshinol suggested that plasma M1 could be a marker to reflect the change in methylation extent and size. The PK and mechanistic disposition data of tanshinol metabolites might be useful for identification of PK markers in the follow-up toxicological study.
In China, Danshen is extensively used for treatment of angina pectoris; patients receiving Danshen therapies may also receive synthetic antianginal agents. Because tanshinol is a key compound in the Danshen therapy, it is important to fully assess the potential for tanshinol-associated herb-drug interactions. In humans, tanshinol is eliminated primarily via renal excretion (mainly involving OAT-mediated tubular secretion) and COMT-mediated methylation. Because of their broad substrate specificity, these drug transporters and metabolizing enzymes are very likely to be targets for tanshinol-associated herb-drug interactions. The study by Jia et al indicated that tanshinol showed low propensity to cause human OAT1/OAT2/OAT3-mediated renal herb-drug interactions and interactions mediated by many other renal transporters28. Like tanshinol, its major metabolite 3-O-methyltanshinol (M1) was also found to have low affinity for OAT1 (Km, 219 μmol/L), OAT2 (2854 μmol/L), and OAT3 (871 μmol/L), suggesting a low potential for M1-perpetrated drug interactions on these renal transporters. Unlike many COMT inhibitors (such as entacapone, tolcapone, and nitecapone; Ki, 0.001–0.1 μmol/L) and dietary catechol-containing polyphenols (such as levo-epigallocatechin-3-gallate, caffeic acid, and chlorogenic acid; IC50, 0.07–3 μmol/L)54,55,56, tanshinol exhibited low affinity for human COMT (Km, 51 μmol/L). In rats, the entacapone-impaired methylation of tanshinol did not significantly influence the systemic exposure to unchanged tanshinol, due to compensation by the sulfation and renal excretion of tanshinol. A similar scenario is expected to take place in humans because the renal excretion of tanshinol is also very extensive, like the renal excretion in rats. These data suggested that tanshinol probably had a low propensity as an inhibitory perpetrator or as a substrate victim to cause COMT-mediated herb-drug interactions at clinically relevant concentrations. In addition, it was also necessary to evaluate possible herb-drug interactions that were perpetrated by tanshinol and M1 on other human drug-metabolizing enzymes and transporters, which are relevant to the elimination of synthetic antianginal agents. Tanshinol and M1 had very little or no inhibitory activities against human P450 enzymes, OATP1B1, OATP1B3, MDR1, MRP2, and BCRP. Taken together, tanshinol and its major human metabolite had low propensity to cause interactions with synthetic antianginal agents.
In summary, methylation, preferentially in the 3-hydroxyl group, was the major metabolic pathway of tanshinol. Tanshinol also underwent considerable sulfation in rats, but this sulfation appeared to be poor in the human liver. The tanshinol metabolites were mainly eliminated via renal excretion, which appeared to mainly involve OAT1-mediated tubular secretion. Methylated tanshinol had no vasodilatory activity; the overall antianginal effect of tanshinol might be attributed primarily to unchanged tanshinol. Entacapone-impaired methylation did not considerably increase systemic exposure to tanshinol in rats. Saturation of tanshinol methylation in the liver could be predicted from the Km value for COMT. Tanshinol and its major human metabolite had low potential for PK interaction with synthetic antianginal agents. Full hazard identification and risk assessment of tanshinol are essential to support long-term use of the Danshen-based antianginal therapy; the current study will help define the risk of hyperhomocysteinemia related to tanshinol methylation. Tanshinol was reported to exhibit endothelial protection against homocysteine-induced injury; the current study will also improve understanding of this pharmacological property of tanshinol.
Author contribution
Chuan LI, Jun-ling YANG, and Dan-dan TIAN designed the research; Dan-dan TIAN, Jun-ling YANG, Wei-wei JIA, Xin-wei LIU, Dan-dan WANG, Jun-hua LIU, Jia-jia DONG, Li LI, Fei-fei DU, Fang XU, Feng-qing WANG, Yan SUN, Yu-xing HUANG, Mei-juan Li, Li-hong HU, Yan ZHU, and Xiu-mei GAO performed the research; Chuan LI, Jun-ling YANG, and Dan-dan TIAN analyzed data; and Chuan LI, Jun-ling YANG, and Dan-dan TIAN wrote the paper.
References
Zhou LM, Zuo Z, Chow MSS . Danshen: an overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. J Clin Pharmacol 2005; 45: 1345–59.
Cheng TO . Cardiovascular effects of Danshen. Int J Cardiol 2007; 121: 9–22.
Jia YL, Huang FY, Zhang SK, Leung SW . Is danshen (Salvia miltiorrhiza) dripping pill more effective than isosorbide dinitrate in treating angina pectoris? A systematic review of randomized controlled trials. Int J Cardiol 2012; 157: 330–40.
Chan K, Chui SH, Wong DYL, Ha WY, Chan CL, Wong RNS . Protective effects of Danshensu from the aqueous extract of Salvia miltiorrhiza (Danshen) against homocysteine-induced endothelial dysfunction. Life Sci 2004; 75: 3157–71.
Lam FF, Yeung JH, Chan KM, Or PM . Relaxant effects of danshen aqueous extract and its constituent danshensu on rat coronary artery are mediated by inhibition of calcium channels. Vascul Pharmacol 2007; 46: 271–7.
Cao YG, Chai JG, Chen YC, Zhao J, Zhou J, Shao JP, et al. Beneficial effects of danshensu, an active component of Salvia miltiorrhiza, on homocysteine metabolism via the trans-sulphuration pathway in rats. Br J Pharmacol 2009; 157: 482–90.
Yang RX, Huang SY, Yan FF, Lu XT, Xing YF, Liu Y, et al. Danshensu protects vascular endothelia in a rat model of hyperhomocysteinemia. Acta Pharmacol Sin 2010; 31: 1395–400.
Zhou X, Chan SW, Tseng HL, Deng Y, Hoi PM, Choi PS, et al. Danshensu is the major marker for the antioxidant and vasorelaxation effects of Danshen (Salvia miltiorrhiza) water-extracts produced by different heat water-extractions. Phytomedicine 2012; 19: 1263–9.
Wang DD, Fan GW, Wang YF, Liu HT, Wang BY, Dong J, et al. Vascular reactivity screen of Chinese medicine danhong injection identifies Danshensu as a NO-independent but PGI2-mediated relaxation factor. J Cardiovasc Pharmacol 2013; 62: 457–65.
Han JY, Fan JY, Horie Y, Miura S, Cui DH, Ishii H, et al. Ameliorating effects of compounds derived from Salvia miltiorrhiza root extract on microcirculatory disturbance and target organ injury by ischemia and reperfusion. Pharmacol Ther 2008; 117: 280–95.
Wang F, Liu YY, Liu LY, Zeng QJ, Wang CS, Sun K, et al. The attenuation effect of 3,4-dihydroxy-phenyl lactic acid and salvianolic acid B on venular thrombosis induced in rat mesentery by photochemical reaction. Clin Hemorheol Microcirc 2009; 42: 7–18.
Tang YQ, Wang MH, Chen CL, Le XY, Sun SJ, Yin YM . Cardiovascular protection with danshensu in spontaneously hypertensive rats. Biol Pharm Bull 2011; 34: 1596–601.
Lu T, Yang JL, Gao XM, Chen P, Du FF, Sun Y, et al. Plasma and urinary tanshinol from Salvia miltiorrhiza (Danshen) can be used as pharmacokinetic markers for cardiotonic pills, a cardiovascular herbal medicine. Drug Metab Dispos 2008; 36: 1578–86.
Gao YL, Liu ZF, Li GS, Li CM, Li M, Li BF . Acute and subchronic toxicity of danshensu in mice and rats. Toxicol Mech Methods 2009; 19: 363–8.
Li GS, Gao YL, Li SJ, Li CM, Zhu XY, Li M, et al. Study on toxicity of danshensu in beagle dogs after 3-month continuous intravenous infusion. Toxicol Mech Methods 2009; 19: 441–6.
Zhang ZC, Xu M, Sun SF, Qiao X, Wang BR, Han J, et al. Metabolic analysis of four phenolic acids in rat by liquid chromatography-tandem mass spectrometry. J Chromatogr B 2008; 871: 7–14.
Brosnan JT, Brosnan ME . The sulfur-containing amino acids: an overview. J Nutr 2006; 136: 1636S–40S.
McCully KS . Homocysteine, vitamins, and vascular disease prevention. Am J Clin Nutr 2007; 86: 1563S–8S.
Smith AD, Smith SM, de Jager CA, Whitbread P, Johnston C, Agacinski G, et al. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: a randomized controlled trial. PLoS ONE 2010; 5: e 12244.
Zhu BT . Catechol-O-methyltransferase (COMT)-mediated methylation metabolism of endogenous bioactive catechols and modulation by endobiotics and xenobiotics: importance in pathophysiology and pathogenesis. Curr Drug Metab 2002; 3: 321–49.
Stead LM, Jacobs RL, Brosnan ME, Brosnan JT . Methylation demand and homocysteine metabolism. Adv Enzyme Regul 2004; 44: 321–33.
Kuhn W, Roebroek R, Blom H, van Oppenraaij D, Müller T . Hyperhomocysteinaemia in Parkinson's disease. J Neurol 1998; 245: 811–2.
Müller T, Werne B, Fowler B, Kuhn W . Nigral endothelial dysfunction, homocysteine, and Parkinson's disease. Lancet 1999; 354: 126–7.
Miller JW, Selhub J, Nadeau MR, Thomas CA, Feldman RG, Wolf PA . Effect of L-dopa on plasma homocysteine in PD patients, relationship to B-vitamin status. Neurology 2003; 60: 1125–9.
Chen F, Li L, Xu F, Sun Y, Du FF, Ma XT, et al. Systemic and cerebral exposure to and pharmacokinetics of flavonols and terpene lactones after dosing standardized Ginkgo biloba leaf extracts to rats via different routes of administration. Br J Pharmacol 2013; 170: 440–57.
Cheng C, Liu XW, Du FF, Li MJ, Xu F, Wang FQ, et al. Sensitive assay for measurement of volatile borneol, isoborneol, and the metabolite camphor in rat pharmacokinetic study of Borneolum (Bingpian) and Borneolum syntheticum (synthetic Bingpian). Acta Pharmacol Sin 2013; 34: 1337–48.
Reagan-Shaw S, Nihal M, Ahmad N . Dose translation from animal to human studies revisited. FASEB J 2008; 22: 659–61.
Jia WW, Du FF, Liu XW, Jiang RR, Xu F, Yang JL, et al. Renal tubular secretion of tanshinol: molecular mechanism, impact on its systemic exposure, and propensity for dose-related nephrotoxicity and renal herb-drug interactions. Drug Metab Dispos 2015; 43: 669–78.
Li L, Zhao YS, Du FF, Yang JL, Xu F, Niu W, et al. Intestinal absorption and presystemic elimination of various chemical constituents present in GBE50 extract, a standardized extract of Ginkgo biloba leaves. Curr Drug Metab 2012; 13: 494–509.
Yang JL, He MM, Niu W, Wrighton SA, Li L, Liu Y, et al. Metabolic capabilities of cytochrome P450 enzymes in Chinese liver microsomes compared with those in Caucasian liver microsomes. Br J Clin Pharmacol 2012; 73: 268–84.
Jiang RR, Dong JJ, Li XX, Du FF, Jia WW, Xu F, et al. Molecular mechanisms governing differential pharmacokinetics of ginsenosides and potential for ginsenoside-perpetrated herb-drug interactions on human OATP1B3. Br J Pharmacol 2015; 172: 1059–73.
Yasuda S, Yasuda T, Hui Y, Liu MY, Suiko M, Sakakibara Y, et al. Concerted action of the cytosolic sulfotransferase, SULT1A3, and catechol-O-methyltransferase in the metabolism of dopamine in SK-N-MC human neuroblastoma cells. Neurosci Res 2009; 64: 273–9.
Kurogi K, Alazizi A, Liu MY, Sakakibara Y, Suiko M, Sugahara T, et al. Concerted actions of the catechol-O-methyltransferase and the cytosolic sulfotransferase SULT1A3 in the metabolism of catecholic drugs. Biochem Pharmacol 2012; 84: 1186–95.
Riches Z, Stanley EL, Bloomer JC, Coughtrie MWH . Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTs) in human tissues: the SULT “pie”. Drug Metab Dispos 2009; 37: 2255–61 .
Guo B, Li C, Wang GJ, Chen LS . Rapid and direct measurement of free concentrations of highly protein-bound fluoxetine and its metabolite norfluxetine in plasma. Rapid Commun Mass Spectrom 2006; 20: 39–47.
Isoherranen N, Hachad H, Yeung CK, Levy RH . Qualitative analysis of the role of metabolites in inhibitory drug-drug interactions: literature evaluation based on the metabolism and transport drug interaction database. Chem Res Toxicol 2009; 22: 294–8.
Yeung CK, Fujioka Y, Hachad H, Levy RH, Isoherranen N . Are circulating metabolites important in drug-drug interaction? Quantitative analysis of risk prediction and inhibitory potency. Clin Pharmacol Ther 2011; 89: 105–13.
Loi CM, Smith DA, Dalvie D . Which metabolites circulate? Drug Metab Dispos 2013; 41: 933–51.
Yu HB, Tweedie D . A perspective on the contribution of metabolites to drug-drug interaction potential: the need to consider both circulating levels and inhibition potency. Drug Metab Dispos 2013; 41: 536–40.
Zamek-Gliszczynski MJ, Chu XY, Polli JW, Paine MF, Galetin A . Understanding the transport properties of metabolites: case studies and considerations for drug development. Drug Metab Dispos 2014; 42: 650–64.
Michel T . Treatment of myocardial ischemia. In: Brunton LL, Lazo JS, Parker KL, editors Goodman & Gilman's The Pharmacological Basis of Therapeutics. McGraw-Hill, New York; 2006. p 823–44.
Tachjian A, Maria V, Jahangir A . Use of herbal products and potential interactions in patients with cardiovascular diseases. J Am Coll Cardiol 2010; 55: 515–25.
Scheen AJ . Cytochrome P450-mediated cardiovascular drug interactions. Expert Opin Drug Metab Toxicol 2011; 7: 1065–82.
Forester SC, Lambert JD . Synergistic inhibition of lung cancer cell lines by (–)-epigallocatechin-3-gallate in combination with clinically used nitrocatechol inhibitors of catechol-O-methyltransferase. Carcinogenesis 2014; 35: 365–72.
Singh R and Hu M . Drug metabolism in gastrointestinal tract. In: Hu M, Li XL, editors. Oral bioavailability: basic principles, advanced concepts, and applications. Hoboken (NJ, USA): John Wiley & Sons, Inc; 2011. p 129–39.
Bonifati V, Meco G . New, selective catechol-O-methyltransferase inhibitors as therapeutic agents in Parkinson's disease. Pharmacol Ther 1999; 81: 1–36.
Li MJ, Wang FQ, Huang YH, Du FF, Zhong CC, Olaleye OE, et al. Systemic exposure to and disposition of catechols, derived from Salvia miltiorrhiza roots (Danshen), after intravenous administration of DanHong injection in human subjects, rats, and dogs. Drug Metab Dispos 2015; 43: 679–90.
O'Flynn N, Timmis A, Henderson R, Rajesh S, Fenu E . Management of stable angina: summary of NICE guidance. Br Med J 2011; 343: d4147.
Siama K, Tousoulis D, Papageorgiou N, Siasos G, Tsiamis E, Bakogiannis C, et al. Stable angina pectoris: current medical treatment. Curr Pharm Des 2013; 19: 1569–80.
Dierkes J, Westphal S . Effect of drugs on homocysteine concentrations. Semin Vasc Med 2005; 5: 124–39.
Lundström K, Salminen M, Jalanko A, Savolainen R, Ulmanen I . Cloning and characterization of human placental catechol-O-methyltransferase cDNA. DNA Cell Biol 1991; 10: 181–9.
Daly D, Miller JW, Nadeau MR, Selhub J . The effect of L-dopa administration and folate deficiency on plasma homocysteine concentrations in rats. J Nutr Biochem 1997; 8: 634–40.
Nissinen E, Nissinen H, Larjonmaa H, Väänänen A, Helkamaa T, Reenilä I, et al. The COMT inhibitor, entacapone, reduces levodopa-induced elevations in plasma homocysteine in healthy adult rats. J Neural Transm 2005; 112: 1213–21.
Männistö PT, KaaKKola S . Catechol-O-methyltransferase (COMT): biochemistry molecular biology, pharmacology, and clinical effecacy of the new selective COMT inhibitors. Pharmacol Rev 1999; 51: 593–628.
Chen DP, Wang CY, Lambert JD, Ai N, Welsh WJ, Yang CS . Inhibition of human liver catechol-O-methyltransferase by tea catechins and their metabolites: structure-activity relationship and molecular-modeling studies. Biochem Pharmacol 2005; 69: 1523–31.
Lee WJ, Zhu BT . Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis 2006; 27: 269–77.
Acknowledgements
This work was funded by grants from the National Natural Science Foundation of China for Distinguished Young Scholars (30925044), the National Science & Technology Major Project of China ′Key New Drug Creation and Manufacturing Program′ (2009ZX09304-002) and the National Basic Research Program of China (2012CB518403). This work was presented in part as a poster presentation at the 10th International ISSX Meeting, Toronto, Canada, September 29 – October 3, 2013: Yang JL et al, Metabolism of and its impact on systemic exposure to tanshinol, a vasoactive compound from of Salvia miltiorrhiza roots (Danshen), in rats.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information is available at website of Acta Pharmacologica Sinica. Figure S1 LC-MS chromatograms and MSE spectra of tanshinol and its metabolites.
Supplementary information
Supplementary Information, Figure S1
LC-MS chromatograms and MSE spectra of tanshinol and its metabolites. (PDF 1581 kb)
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Tian, Dd., Jia, Ww., Liu, Xw. et al. Methylation and its role in the disposition of tanshinol, a cardiovascular carboxylic catechol from Salvia miltiorrhiza roots (Danshen). Acta Pharmacol Sin 36, 627–643 (2015). https://doi.org/10.1038/aps.2015.20
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2015.20
Keywords
This article is cited by
-
Pharmacologically significant constituents collectively responsible for anti-sepsis action of XueBiJing, a Chinese herb-based intravenous formulation
Acta Pharmacologica Sinica (2024)
-
Pharmacokinetics-based identification of pseudoaldosterogenic compounds originating from Glycyrrhiza uralensis roots (Gancao) after dosing LianhuaQingwen capsule
Acta Pharmacologica Sinica (2021)
