
Abstract
There is accumulating evidence that aggregating, misfolded proteins may have an impact on autophagic function, suggesting that this could be a secondary pathological mechanism in many diseases. In this review, we focus on the role of autophagy in four major neurodegenerative diseases: Alzheimer disease (AD), Huntington's disease (HD), Parkinson's disease (PD) and amyotropic lateral sclerosis.
Similar content being viewed by others
Introduction to autophagy
Macroautophagy (which we will call autophagy) is initiated by the formation of a cup-shaped double-membrane structure (the phagophore) in the cytoplasm. The origin of this structure is still under investigation, but currently the endoplasmic reticulum, Golgi, mitochondria and the plasma membrane are all proposed to be potential sources (which may not be mutually exclusive)1,2,3. The phagophore edges expand and then seal to engulf intracytoplasmic cargo, such as protein oligomers, organelles and ribosomes, thereby sequestering the cargo in a double-membrane called an autophagosome. Autophagosomes are then trafficked along microtubules towards the microtubule-organizing centre, where they mature through fusion with multivesicular bodies and early and/or late endosomes, before fusing with lysosomes. The autophagosomal contents are then degraded by lysosomal hydrolases and the degradation products are then transported back into the cytoplasm to be recycled.
It has been well established that autophagy regulates important biological functions, such as cell survival, cell death, cell metabolism, development, aging, infection and immunity. At a cellular level, the involvement of autophagy in the cell death and cell survival processes appears to be complex. The visualization of autophagosomes in dying cells has led certain groups to conclude that autophagy can serve as a nonapoptotic form of programmed cell death4. Although cells can manifest a clear increase in the numbers of autophagosomes shortly before or during their death, this phenomenon is sometimes due to defects in autophagosomal maturation and, hence, decreased, rather than increased, autophagy5,6. Most evidence indicates that autophagy is primarily a pro-survival rather than a pro-death mechanism, and in the context of neurodegenerative disorders, an emerging consensus is that induction of autophagy is a neuroprotective response and that defective autophagy promotes pathology.
Autophagy malfunction and neurodegenerative diseases
There is accumulating evidence that aggregating, misfolded proteins may have an impact on autophagic function, suggesting that this could be a secondary pathological mechanism in many diseases. In this review, we focus on the role of autophagy in four major neurodegenerative diseases: Alzheimer Disease (AD), Huntington's Disease (HD), Parkinson's Disease (PD) and Amyotropic Lateral Sclerosis (ALS).
Huntington's disease
Huntington's Disease (HD) is an autosomal dominant disorder caused by the expansion of the polyglutamine repeat in the N-terminus of the Huntington gene. The mutant protein is aggregate-prone and forms many extra-nuclear inclusions in the typical adult-onset case. (In the rarer juvenile-onset cases, inclusions can form in the nucleus). The first clues about the ability of autophagy to influence the accumulation and toxicity of aggregate-prone intracytoplasmic proteins associated with neurodegeneration were made in cell-based models of Huntington's disease, which showed that chemical inhibition of autophagy slowed the clearance of these proteins and enhanced their toxicity, while autophagy induction enhanced mutant protein clearance and was protective7. Interestingly, the mutant protein has a high dependence on autophagy for its clearance, while the wild-type protein turnover is hardly affected by autophagy. This phenomenon may be due to the fact that oligomeric forms of the mutant protein are not accessible to the proteasome, and so need to be cleared via autophagy. On the other hand, the wild-type protein can be very effectively and rapidly cleared by the ubiquitin-proteasome system.
Alzheimer's disease
Post-mortem analysis of AD brains reveal abnormal structures consisting of amyloid plaques and intracellular neurofibrillary tangles comprising hyperphosphorylated protein tau8. Several genetic defects have been identified to cause rare familial forms of AD, such as mutations in amyloid precursor protein (APP) and presenilin (PSEN) 19,10. Within neurons, autophagosomes and endosomes appear to be formed in processes and travel towards lysosomes concentrated in the perinuclear region of the cell body. A prominent feature of AD is the accumulation of autophagosomes, many containing amyloid-peptide. It has been proposed that defects in retrograde transport and therefore impaired vesicle movement within dystrophic neurons, especially those with neurofibrillary tangles may contribute to defective delivery of autophagosomes to lysosomes in Alzheimer's disease11. It is still unclear when this occurs during the course of the condition.
Parkinson's disease
Parkinson's disease is characterized by the selective degeneration of neurons in the substantia nigra and the presence of aggregated α-synuclein-containing intracellular inclusions known as Lewy bodies. Overexpression of wild type α-synuclein is sufficient to cause human PD, as this occurs in families who have duplications of this locus12,13. Excess α-synuclein impairs autophagy in mammalian cells and transgenic mice. Conversely, a reduction in α-synuclein levels enhances autophagy in cells and in mice14,15. Experiments using inhibitors and activators of autophagy confirm that wild–type α-synuclein is degraded by this pathway16,17 and in an α-synuclein transgenic mouse model, delivery of a Beclin-1 encoding lentivirus that induces autophagy ameliorates the synaptic and dendritic pathology and decreases α-synuclein accumulation in the limbic system18.
Amyotropic lateral sclerosis (ALS)
ALS is an adult onset neurodegenerative disease involving selective death of motor neurons in the brain and spinal cord19. Mitochondrial damage and abnormal protein inclusions such as Lewy bodies20, Skein inclusions21 and Bunina inclusions22 are the characteristic pathological features in ALS. ALS can be caused by mutations in a number of different genes. Mutations in DCTN1, which encodes for p150, a subunit of dynactin have been implicated in familial ALS23. Since the dynein apparatus is required for transport of autophagosomes to lysosomes, this may explain the accumulation of aggregate-prone proteins in this form of the disease24,25.
A mutation in an ESCRT (Endosomal Sorting Complexes Required for Transport) protein also causes ALS. Cells depleted of ESCRT have inhibited autophagic degradation, due to impaired autophagosome-lysosome fusion, causing accumulation of ubiquitinated aggregates26. This may explain some of the pathology in patients with ALS who have a mutation in this complex. Another protein mutated in ALS is VCP (Valosin-Containing Protein), which also appears to regulate autophagosome removal27. VCP mutant models accumulate autophagosomes, which fail to mature into autolysosomes28.
It is likely that decreases in autophagic flux will contribute to the progression of this disease by enhancing the accumulation of aggregate-prone proteins, dysfunctional mitochondria, and increasing susceptibility to cell death.
Therapeutic implications for up-regulation of autophagy
As many diseases that are caused by intracytoplasmic aggregate-prone proteins are associated with gain-of-function mutations, a viable therapeutic strategy might be to reduce the accumulation of the toxic protein in the cytoplasm. Indeed promoting the clearance of aggregate-prone proteins via pharmacological induction of autophagy has proved to be a useful mechanism for protecting against the toxic effects of these proteins in a range of cell and animal models7,29,30.
Targeting the mTOR-dependent pathway
Chemical induction of autophagy protects cells against the toxic insults of aggregate-prone proteins associated with neurodegeneration by promoting their clearance7 and also by reducing susceptibility to caspase activation and apoptosis31. The first known drug used in people identified as an autophagy inducer was rapamycin, which was already in clinical use for other indications. In mammalian cells, rapamycin inhibits the kinase activity of mTOR by forming a complex with the immunophilin FK506-binding protein of 12 kDa (FKBP12) that binds to and inactivates mTOR, leading to the upregulation of autophagy29,32,33. Rapamycin treatment enhances the clearance of mutant huntingtin fragments, reduces aggregate formation and protects against toxicity in cell, Drosophila and mouse models of Huntington's disease (HD)29,31,34. In cell models, rapamycin promotes the clearance of other intracytoplasmic, disease-associated, aggregate-prone proteins, including certain mutant proteins associated with spinocerebellar ataxias, mutant forms of α-synuclein implicated in PD, and mutant tau responsible for FTD29,35,36. It is likely that autophagy regulates clearance of SDS-soluble species of these proteins, and that the formation of large aggregates visible by light microscopy is influenced by autophagy clearing the smaller aggregate precursors. In Drosophila models of these diseases, the benefits of rapamycin appear to be autophagy-dependent, as this drug had no effects on the proteinopathy toxicity in flies expressing these mutant proteins when the activity of different autophagy genes were reduced29,37,38. Consistent with the Drosophila data, the rapamycin analogue CCI-779 reduces both mutant huntingtin and ataxin-3 levels and ameliorates toxicity in mouse models of HD and spinocerebellar ataxia type 3, respectively39,40.
Targeting the mTOR-independent pathway
The disadvantage of TOR targeting is that its substrates control several cellular processes besides autophagy, including repression of ribosome biogenesis and protein translation29,41. These processes likely contribute to the complications seen with its long-term use. Accordingly, there have been a series of studies to identify novel autophagy-upregulating compounds with the subsequent discovery of pathways that are independent of the target of rapamycin42.
For example, lithium, a mood stabilising drug used for the long-term treatment of affective disorders, facilitates the clearance of mutant huntingtin in HD cell and Drosophila models by reducing IP3 levels, and reduces mutant protein-associated aggregation and toxicity43. Consistent with the role of IP3 in autophagy, pharmacological inhibition of the IP3R by xestospongin B also induces autophagy44. Additionally, sodium valproate and carbamazepine, which inhibit inositol synthesis and therefore decrease IP3 levels, also reduce the accumulation and aggregation of mutant huntingtin and its toxicity in HD cell models, and protect against neurodegeneration in Drosophila models of HD43,45,46. Rilmenidine, an imidazoline receptor 1 agonist, induces autophagy, enhances mutant huntingtin clearance and reduces toxicity in an HD mouse model30. Since this drug is a centrally-acting hypertensive agent with minimal side-effects, it is currently being tested in a safety trial in HD patients.
Future perspectives
For many diseases, the upregulation of autophagy is a promising therapeutic target. Combining knowledge of the potential mechanisms of autophagy compromise in neurodegenerative proteinopathies with knowledge of the range of signalling pathways and drugs available to control autophagy may enhance the development of optimal therapeutics.

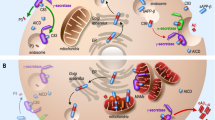
A schematic of autophagy showing stages disrupted in selected neurodegenerative diseases. In starvation conditions, or growth factor deprivation, autophagy is stimulated due to mammalian target of rapamycin (mTOR) inhibition. Autophagy begins with the formation of a cup-shaped double-membrane structure, termed a phagophore. This is followed by membrane elongation and fusion to form a complete autophagosome, engulfing mutant and/or aggregate-prone proteins such as excess alpha-synuclein from the cytoplasm. Autophagosomes are then trafficked along microtubule tracks via dynein motors towards the lysosomes where autophagosome-lysosome fusion occurs, generating an autophagolysosome. Excess alpha-synuclein disrupts autophagic initiation in Parkinson's disease (PD). In amyotropic lateral sclerosis (ALS), mutations in the dynein/dynactin complex inhibit autophagosome trafficking to lysosomes. Inefficient autophagosome-lysosome fusion and defects in lysosomal acidification contributes to pathogenesis in forms of Alzheimer's disease (AD).
References
Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A . A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 2009; 11: 1433–7.
Tooze SA, Yoshimori T . The origin of the autophagosomal membrane. Nat Cell Biol 2010; 12: 831–5.
Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC . Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010; 12: 747–57.
Clarke PG, Puyal J . Autophagic cell death exists. Autophagy 2012; 8: 867–9.
Levine B, Yuan J . Autophagy in cell death: an innocent convict? J Clin Invest 2005; 115: 2679–88.
Kroemer G, Levine B . Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9: 1004–10.
Ravikumar B, Duden R, Rubinsztein DC . Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 2002; 11: 1107–17.
Zerovnik E . Protein conformational pathology in Alzheimer's and other neurodegenerative diseases: new targets for therapy. Curr Alzheimer Res 2010; 7: 74–83.
Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, et al. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature 1992; 360: 672–4.
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med 1996; 2: 864–70.
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010; 141: 1146–58.
Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 2004; 364: 1167–9.
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. Alpha-Synuclein locus triplication causes Parkinson's disease. Science 2003; 302: 841.
Winslow AR, Rubinsztein DC . The Parkinson disease protein alpha-synuclein inhibits autophagy. Autophagy 2010; 7: 429–31.
Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, et al. Alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol 2010; 190: 1023–37.
Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC . Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 2003; 278: 25009–13.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D . Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004; 305: 1292–5.
Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci 2009; 29: 13578–88.
Rowland LP, Shneider NA . Amyotrophic lateral sclerosis. N Engl J Med 2001; 344: 1688–700.
Mather K, Watts FZ, Carroll M, Whitehead P, Swash M, Cairn N, et al. Antibody to an abnormal protein in amyotrophic lateral sclerosis identifies Lewy body-like inclusions in ALS and Lewy bodies in Parkinson's disease. Neurosci Lett 1993; 160: 13–6.
Leigh PN, Anderton BH, Dodson A, Gallo JM, Swash M, Power DM . Ubiquitin deposits in anterior horn cells in motor neurone disease. Neurosci Lett 1988; 93: 197–203.
Okamoto K, Hirai S, Amari M, Watanabe M, Sakurai A . Bunina bodies in amyotrophic lateral sclerosis immunostained with rabbit anti-cystatin C serum. Neurosci Lett 1993; 162: 125–8.
Münch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004; 63: 724–6.
Jahreiss L, Menzies FM, Rubinsztein DC . The itinerary of autophagosomes: From peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008; 9: 574–87.
Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O'Kane CJ, et al. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet 2005; 37: 771–6.
Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerød L, Fisher EM, et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 2007; 179: 485–500.
Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010; 68: 857–64.
Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 2009; 187: 875–88.
Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet 2006; 15: 433–42.
Rose C, Menzies FM, Renna M, Acevedo–Arozena A, Corrochano S, Sadiq O, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington's disease. Hum Mol Genet 2010; 19: 2144–53.
Ravikumar B, Berger Z, Vacher C, O'Kane CJ, Rubinsztein DC . Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet 2006; 15: 1209–16.
Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002; 110: 163–75.
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006; 22: 159–68.
Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC . Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 2009; 16: 46–56.
Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, et al. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 2006; 76: 89–101.
Menzies FM, Ravikumar B, Rubinsztein DC . Protective roles for induction of autophagy in multiple proteinopathies. Autophagy 2006; 2: 224–5.
Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007; 447: 859–63.
Wang T, Lao U, Edgar BA . TOR-mediated autophagy regulates cell death in Drosophila neurodegenerative disease. J Cell Biol 2009; 186: 703–11.
Menzies FM, Huebener J, Renna M, Bonin M, Riess O, Rubinsztein DC . Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 2010; 133: 93–104.
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36: 585–95.
Yang YP, Liang ZQ, Gu ZL, Qin ZH . Molecular mechanism and regulation of autophagy. Acta Pharmacol Sin 2005; 26: 1421–34.
Fleming A, Noda T, Yoshimori T, Rubinsztein DC . Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol 2011; 7: 9–17.
Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170: 1101–11.
Wong A, Grubb DR, Cooley N, Luo J, Woodcock EA . Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 2007; 14: 1029–39.
Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008; 4: 295–305.
Sarkar S, Rubinsztein DC . Small molecule enhancers of autophagy for neurodegenerative diseases. Mol Biosyst 2008; 4: 895–901.
Acknowledgements
DCR is funded by a Wellcome Trust Principal Fellowship, a Wellcome Trust/MRC Strategic Grant on Alzheimer's disease, the Tau Consortium, and the Biomedical Research Unit in Dementia at Addenbrooke's Hospital. We are grateful to HM's Government of Brunei Darussalam (SHFL), The Oppenheimer Memorial Trust, The Cambridge Commonwealth Trust and The Higher Education Funding Council for England (WEH).
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Hochfeld, W., Lee, S. & Rubinsztein, D. Therapeutic induction of autophagy to modulate neurodegenerative disease progression. Acta Pharmacol Sin 34, 600–604 (2013). https://doi.org/10.1038/aps.2012.189
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2012.189
Keywords
This article is cited by
-
Autophagy in trimethyltin-induced neurodegeneration
Journal of Neural Transmission (2020)
-
Mechanisms of protein toxicity in neurodegenerative diseases
Cellular and Molecular Life Sciences (2018)
-
Small molecule LX2343 ameliorates cognitive deficits in AD model mice by targeting both amyloid β production and clearance
Acta Pharmacologica Sinica (2016)
-
Cannabidiol Post-Treatment Alleviates Rat Epileptic-Related Behaviors and Activates Hippocampal Cell Autophagy Pathway Along with Antioxidant Defense in Chronic Phase of Pilocarpine-Induced Seizure
Journal of Molecular Neuroscience (2016)
-
Alzheimer's disease progression model based on integrated biomarkers and clinical measures
Acta Pharmacologica Sinica (2014)
