
Abstract
Polynoid scale worms (Polynoidae, Annelida) invaded deep-sea chemosynthesis-based ecosystems approximately 60 million years ago, but little is known about their genetic adaptation to the extreme deep-sea environment. In this study, we reported the first two transcriptomes of deep-sea polynoids (Branchipolynoe pettiboneae, Lepidonotopodium sp.) and compared them with the transcriptome of a shallow-water polynoid (Harmothoe imbricata). We determined codon and amino acid usage, positive selected genes, highly expressed genes and putative duplicated genes. Transcriptome assembly produced 98,806 to 225,709 contigs in the three species. There were more positively charged amino acids (i.e., histidine and arginine) and less negatively charged amino acids (i.e., aspartic acid and glutamic acid) in the deep-sea species. There were 120 genes showing clear evidence of positive selection. Among the 10% most highly expressed genes, there were more hemoglobin genes with high expression levels in both deep-sea species. The duplicated genes related to DNA recombination and metabolism, and gene expression were only enriched in deep-sea species. Deep-sea scale worms adopted two strategies of adaptation to hypoxia in the chemosynthesis-based habitats (i.e., rapid evolution of tetra-domain hemoglobin in Branchipolynoe or high expression of single-domain hemoglobin in Lepidonotopodium sp.).
Similar content being viewed by others


Introduction
The discoveries of hydrothermal vents in the late 1970s and cold seeps in the early 1980s have revolutionized our view of life in the deep sea. Hydrothermal vents discharge geothermally heated water rich in methane, hydrogen sulfide and other hydrocarbon-rich fluids, while cold seeps release the same dissolved organic chemicals slowly without causing an appreciable temperature rise1. Despite the differences in the source of reduced organic compounds and temperature in the fluids, hydrothermal vents and cold seeps are both productive ecosystems in the deep sea2, and they can share more than 20% species when occur in the same region, indicating their genetic connectivity3.
There has been great interest in understanding how animals have adapted to the extreme conditions in these habitats that are characterized by high pressure, darkness, variable temperatures and high concentrations of toxic substances4,5,6,7,8,9. However, except for few species10,11, the limited genomic resources have hindered studies of physiological adaptation at the molecular level for animals living in these extreme environments.
Advances in sequencing technologies over the last few years have provided unprecedented opportunities for biologists. To date, these technologies have been applied in various areas of genomic research including whole genome sequencing, whole transcriptome sequencing, targeted resequencing, and noncoding RNA sequencing. Among these research areas, transcriptome sequencing, which targets the expressed gene transcripts, has been applied widely to gene discovery and to understanding the genetic base of adaptation to extreme environments due to its high affordability12,13. For instance, transcriptome sequencing was applied to analyze gene duplication in an Antarctic icefish, and found that the duplicated genes are highly enriched in mitochondrial functions14. It was used to understand the adaptation of a highland fish, which showed that genes related to hypoxia and energy metabolism have undergone rapid evolution15. This technique was also applied to analyze several depth-related genes (LDH-A, LDH-B, MDHc, ACTA1 isoform 1) in a deep-sea fish which indicated that these genes are constrained by purifying selection16. Moreover, transcriptome analysis of a hydrothermal vent annelid showed that high frequency of charged versus polar residues can be responsible for its thermo-adaptation17.
Scale worms in the family Polynoidae are well represented from the shallow intertidal to the deep sea18,19,20. In the deep sea, more than 120 species in 53 genera of polynoids have been reported from various chemosynthesis-based habitats, including hydrothermal vents, cold seeps, sunken wood and whale carcasses21,22. These scale worms are believed to be the descendants of shallow-water species that invaded the deep sea ca. 60 million years ago, and thereafter diverged to colonize various chemosynthesis-based habitats across the world’s oceans23. Members of the genus Branchipolynoe (Polynoidae) are commonly found in the mantle cavity of Bathymodiolus mussels living in hydrothermal vents and cold seeps24,25. Currently this genus has three species described from different biogeographic areas26: B. symmytilida from the hydrothermal vents of the Galápagos Rift in the eastern Pacific27, B. seepensis from the cold-seeps of the Florida Escarpment in the Gulf of Mexico28, and B. pettiboneae from the hydrothermal vents of the Okinawa Trough and the Kaikata Seamount in the northwestern Pacific29. Undescribed species of Branchipolynoe have been reported in other regions, such as the Central Indian Ridge1 and sunken vegetation off Papua New Guinea30. The widespread geographic occurrence of Branchipolynoe spp. makes them an excellent model in studies of population genetics, speciation, and biogeography31,32. Moreover, previous studies have shown that Branchipolynoe spp. have some morphological and molecular adaptations that have allowed them to thrive in the deep-sea chemosynthesis habitats that often experience hypoxia33,34, making them an excellent model for studies of adaptive evolution. Specifically, their parapodia bear well-developed arborescent branchiae, which are used to facilitate gaseous exchange with the environment35. Their coelomic fluid is red, containing high concentrations of hemoglobin for oxygen storage and transport. These morphological and molecular adaptations are believed to be essential for their survival in the chemosynthesis habitats that are often hypoxic, with oxygen concentrations being less than 2 mg/L1,36,37. The hemoglobins in B. symmytilida and B. seepensis have high affinity to oxygen; it has a tetra-domain structure that is unique among the hemoglobins of polychaetes23,34. Lepidonotopodium spp. are free-living polynoids in deep-sea hydrothermal vents21. They do not bear branchiae but their coelomic fluid is also red and contains a one-domain hemoglobin23.
To gain insight into the genetic basis of adaptation of deep-sea polynoids to extreme environmental conditions, we sequenced the transcriptomes of B. pettiboneae and Lepidonotopodium sp., and compared them with that of the shallow-water polynoid Harmothoe imbricata, which is in a clade of polynoids that is closest in phylogenetic relationship with all deep-sea polynoids20. Harmothoe imbricata is commonly found in low intertidal and subtidal rocky shores of North America and northern Europe37. We aim to provide genomic resources for the deep-sea polynoids, and discover several aspects of their transcriptomic differences with the shallow-water species, including codon and amino acid usage, expression of highly expressed genes, composition of duplicated genes, as well as evolutionary patterns of selected genes that may have undergone positive selection. In a broader sense, the findings will help us better understand how animals have adapted to deep-sea extreme environments.
Materials and Methods
Obtaining high-throughput cDNA sequences
Specimens of Branhipolynoe pettiboneae were collected from Jiaolong Ridge, a cold seep in the South China Sea (22°06.921′N, 119°17.131′E, depth 1122 m) in June 2013 during the first scientific cruise of the manned submersible Jiaolong. At this site, B. pettiboneae lived inside the mantle cavity of the deep-sea mussel Bathymodiolus platifons. The site temperature was 3.6 °C, but dissolved oxygen concentration was not measured. Specimens of Lepidonotopodium sp. were collected from the Noho site, a hydrothermal vent in the Okinawa Trough (27°31.0′N, 126°58.9′E, depth 1550 m), using ROV Kaiko MK-IV on-board R/V Kairei during the cruise KR15-17 in November 2015. The site temperature was 3.84 °C, and dissolved oxygen concentration was 1.76 mg/L.
The samples were kept in partially insulated sample boxes, and were preserved in RNALater (Ambion, Austin, TX) immediately once they were brought to the deck, which was roughly three hours after sampling. The samples were later transported to Hong Kong Baptist University on dry ice. For both deep-sea species, total RNA was extracted from three middle parapodia with elytra of a specimen using TRIzol reagent (Invitrogen, Carsbad, NM, USA) following the manufacturer’s protocol. The quality of RNA samples were detected by 1% agarose gel electrophoresis and stained using GelRed (Biotium, Fremont, CA, USA), then visualized under ultraviolet light. The quantity of RNA samples were analyzed using Bioanalyzer 2100 (Agilent Technologies, CA, USA). The RNA samples were sent to Beijing Genomics Institute (BGI), Shenzhen for messenger RNA enrichment, RNA fragmentation, cDNA synthesis, adapter ligation, and PCR amplification38, and the PCR product was sequenced to produce paired-end reads of 100 bp read length for B. pettiboneae using an Illumina Hiseq 2000 in 2014, and 150 bp read length for Lepidonotopodium sp. using an Illumina Hiseq 4000 in 2015. The raw transcriptomic data for Branchipolynoe pettiboneae and Lepidonotopodium sp. are deposited in NCBI SRA database with accession number of SRR4419842 and SRR4419843, respectively. Raw transcriptome sequences of Harmothoe imbricata were obtained from NCBI’s SRA database39 (accession no. SRX1015591). The RNA of this sample was extracted from an elytra of a middle parapodia collected from the intertidal zone of a rocky shore at Land’s End (43.807 °N, 69.995 °W) on Bailey Island, Maine, USA39. Below the rocky shore is a sandy beach and the seawater at the site is well-oxygenated.
Transcriptome assembly and annotation
The raw reads were filtered using Trimmomatic v0.3340. In brief, adapters were removed based on information provided in the TruSeq RNA Library Prep Kit which was used in RNA library preparation (B. pettiboneae and H. imbricata: ILLUMINACLIP:TruSeq2-PE.fa:2:30:10; Lepidonotopodium sp.: ILLUMINACLIP:TruSeq3-PE.fa:2:30:10), the leading and trailing low quality bases were removed (LEADING:3 TRAILING:3), and cut when the average quality per base was below 15 with a 4-base wide sliding window, and removed when the length was below 36 bases (SLIDINGWINDOW:4:15 MINLEN:36). Trimmed paired-end reads of the three species were used for assembly using Trinity trinityrnaseq-2.0.641. RSEM 1.2.1942 was used to estimate the transcript abundance, and the lowly expressed transcript isoforms (IsoPct < 1%) were removed. TransDecoder was then used to identify the candidate open reading frames (ORFs) and peptides in the filtered transcripts. Identical sequences (100% similarity) in predicted peptides were removed by CD-HIT43. The longest predicted protein of each contig was selected using a python script (Supplementary Information, PickUpLongest.py). Annotation was carried out by InterProScan-5.13-52.044. A Gene Ontology (GO) annotation plot was made using WEGO45.
Calculation of GC content and codon usage
The overall GC content and codon usage of transcriptome from B. pettiboneae, Lepidonotopodium sp. and H. imbricata were calculated using codonW 1.4.446. Relative synonymous codon usage (RSCU) was used to indicate the codon usage pattern. RSCU = 1 indicates that the codons are used equally and randomly. RSCU > 1 and RSCU < 1 indicates positive and negative codon usage bias, respectively. Codon usage bias was considered strong when absolute ΔRSCU > 0.147. The proportion of each amino acid is the summation of all its codons.
Identification of orthologous genes and Ka/Ks analysis
Orthologs were identified by pair-wise comparison using OrthoMCL48. The predicted protein sequences with high identity ( > 95%) were removed with CD-HIT to eliminate isoforms caused by alternative splicing. Default settings were used for the OrthoMCL analysis and aligned sequences with less than 50 amino acids were excluded. Only 1:1 orthologous genes were used for base substitution analysis. The input files for Ka/Ks_Calculator49 were prepared using ParaAT1.050. A maximum likelihood method with the MLPB model was used to calculate non-synonymous substitutions per nonsynonymous site (Ka), synonymous substitutions per synonymous site (Ks), and the ratio between Ka and Ks (Ka/Ks). Only ortholog pairs with P-value (Fisher’s exact test) <0.05, 0.01 < Ks < 1, and Ka < 1 were retained51. Ka/Ka > 1 was considered as a clear signal of positive selection. Positively selected genes were annotated based on the GO annotation and InterPro online database52.
Comparison of highly expressed genes in three species
Gene expression level was estimated using RSEM 1.2.19. Based on the reads per kilobase of exon per million reads mapped (FPKM), only highly expressed annotated genes (top 10%) were included in the comparison. The 10% highly expressed genes were classified based on their functional annotation. Their percentage and average expression level were compared to illustrate the differences in cellular processes among the three species.
Identification of putatively duplicated genes
Putatively duplicated genes were identified using OrthoMCL based on an E value of 1e × 10−5 in BLASTP53. Alignments shorter than 50 amino acid residues (i.e. <150 nt) were removed. Pairs of peptide sequences with similarity between 70% and 98% were considered as duplicated genes14. Only clusters with annotation were included in the comparison. Gene Ontology classification was made using WEGO. Functional enrichment analysis was done using GOEAST54. Specifically, the Benjamini and Yekutieli false discovery rate correction under dependency was applied. GO terms with p < 0.05 were considered as significantly enriched based on a hypergeometric statistical test.
Phylogenetic and evolutionary analyses of hemoglobins
To illustrate the use of the transcriptome data, we conducted phylogenetic and evolutionary analyses of hemoglobins in deep-sea scale worms. This group of proteins function in oxygen binding, and could be important in the adaptation of deep-sea scale worms to the hypoxic conditions in chemosynthesis-based ecosystems5,23. Potential hemoglobin sequences were fetched from the assembled transcriptome of the three scale worms using BLASTP with an E value of 1e × 10−5. Hemoglobin sequences from other deep-sea scale worms were also obtained from GenBank. Phylogenetic analyses were conducted using the Maximum Parsimony (MP) and the Maximum Likelihood (ML) methods. A total of 18 hemoglobin sequences from the other deep-sea scale worms including B. seepensis and B. symmytilida obtained from GenBank were added in the alignment and phylogenetic analysis. Several intercellular hemoglobin sequences from Glycera dibranchiata (family Glyceridae), and extracellular hemoglobin sequences from Lumbricus terrestris (family Lumbricidae, Oligochaeta) were used as outgroups. Accession numbers for the hemoglobin sequences are listed in Supplementary Table S1. All the sequences were aligned using Muscle3.8.31 implemented in Mesquite55. The MP tree was constructed using PAUP* V4.056. The ML analysis was conducted using RaXML GUI1.357 with the Dayhoff +I+G model which was determined using ProtTest-3.058. The bootstrap values were all based on 1000 iterations. The candidate single domain and tetra-domain sequences of B. pettiboneae were confirmed based on their similarities with characterized hemoglobins obtained from the phylogenetic analysis. The hemoglobin sequences were analyzed for positive selection using PAML 4.8 following a previous study34.
Results
Transcriptome assembly and annotation
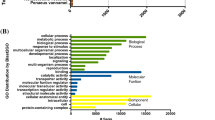
There are 3.8 Gb raw reads in B. pettiboneae, 7.0 Gb raw reads in Lepidonotopodium sp., and 3.9 Gb raw reads in Harmothoe imbricata (Table 1). Removing adaptors and low-quality reads resulted in the retention of 98.4%, 84.7% and 91.3% reads for the three species, respectively. The cleaned reads were used for the assembly and generated 153,339 contigs for B. pettiboneae, 225,709 contigs for Lepidonotopodium sp. and 98,806 contigs for H. imbricata. The assembled transcriptome had an average contig length of 521 to 738 bp with N50 of 583 to 1,198 in the three species. Contig length distributions for the three species are shown in Fig. 1. Short contigs (<1,000 bp) constituted about one third in the three species, but there were about 8% more long contigs ( > 1,000) in Lepidonotopodium sp. There were 38,734, 79,930 and 36,001 predicted proteins in B. pettiboneae, Lepidonotopodium sp. and H. imbricata, respectively. Applying InterProScan revealed 38.7%, 41.2% and 48.6% annotated genes for B. pettiboneae, Lepidonotopodium sp. and H. imbricata, respectively (Table 1; Supplementary Table S2). A WEGO gene ontology annotation plot (Fig. 2) showed that the compositions of genes in the three species were broadly similar in cellular components, molecular functions and biological processes.

Contig length distribution in the assembled transcriptomes of B. pettiboneae, Lepidonotopodium sp. and H. imbricata.

Genes are grouped into three main functional categories: biological process, cellular component and molecular function. The left abscissa indicates the percentage and right one shows the number of annotated genes. One gene may have match in multiple GO terms.
Comparison of GC content and codon usage
The GC content of the B. pettiboneae, Lepidonotopodium sp. and H. imbricata transcriptome is 52.1%, 53.2% and 52%, respectively. The difference in codon usage ranges from 0 to 5.14‰, and the difference in Relative Synonymous Codon Usage (RSCU) ranges from 0 to 0.24. Most of the codons have very similar proportions and RSCU values among the three species. However, 14 codons have strong negative or positive codon usage bias (absolute ΔRSCU > 0.1) in the deep-sea species compared with the shallow-water species (Supplementary Table S3). More than half of the codons with strong biased ΔRSCU values encode the amino acids glycine, proline and cysteine. The usage pattern of amino acids is listed in Table 2. The species difference in proportion for more than half of the amino acids is less than 0.2%. However, there are large differences ( > 0.4%) for several amino acids between the deep-sea and shallow-water species. Specifically, there are more positively charged amino acids (i.e., histidine and arginine) and less negatively charged amino acids (i.e., aspartic acid and glutamic acid) in the deep-sea species. In addition, there are more non-polar amino acid leucine and less glycine in the deep-sea species.
Positively selected genes
There were 5,494, 6,851 and 7,311 pairs of orthologs between B. pettiboneae and H. imbricata, Lepidonotopodium sp. and H. imbricata, and B. pettiboneae and Lepidonotopodium sp., respectively (Table 3). Filtering reduced the numbers to 1,161, 1,801 and 3,269 for the three pairs, respectively. Base substitution analysis revealed a clear signal of positive selection (Ka/Ks > 1) in 192 orthologs between B. pettiboneae and H. imbricata, 306 orthologs between Lepidonotopodium sp. and H. imbricata, and 416 orthologs between B. pettiboneae and Lepidonotopodium sp. However, only 51, 40 and 29 positive selected genes were annotated in the three pair-wise comparisons, respectively (Table 3 and Supplementary Table S4). These positively selected genes are mainly related to six different functions: cell structure and cell adhesion, energy-related process, chromatin structure and gene expression, signal pathway and immunity, protein metabolism and function, and transport (Fig. 3). Among the positively selective orthologs between B. pettiboneae and H. imbricata, genes involved in protein metabolism, signal pathway, immune, and energy-related process accounted for the largest proportions (64% in total). Between Lepidonotopodium sp. and H. imbricata, the largest proportion of positively selected genes is related to protein metabolism and function (43%), followed by signal pathway and immunity (20.0%). Between the two deep-sea species, more than one third of positively selected genes (38%) are involved in chromatin structure and gene expression, and one fifth are involved in signal pathway and immunity.

Biological functions of positively selected genes in pair-wise comparisons among the three species of scale worms.
Comparison of highly expressed genes
The top 10% highly expressed genes included 1465 genes in B. pettiboneae, 3215 genes in Lepidonotopodium sp. and 1733 genes in H. imbricata. These genes had FPKM value ranging from 7.78 to 4527.82 (average 72.8), from 8.77 to 60511.8 (average 117.9) and from 10.22 to 2897.81 (average 80.7) in B. pettiboneae, Lepidonotopodium sp. and H. imbricata, respectively (Supplementary Table S5). Gene groups related to conserved biological processes are compared with respect to their percentage (Fig. 4a) and expression level (Fig. 4b). Gene groups related to several biological processes, including dehydrogenase, transcription, translation, proteolysis and cytoskeleton (actin and tubulin), were of comparable expression levels in the three species. Nevertheless, there were notable differences in the expression of several gene groups among the three species. In particular, globin genes in both deep-sea species were more abundant (about three times) and had higher expression levels (more than ten times) than in the shallow-water species. Gene groups involved in DNA recombination and integration including reverse transcriptase, DNA breaking-rejoining domains and integrase were only present and highly expressed in the two deep-sea species (Fig. 4). Zinc finger and EF hand, which are common protein-DNA binding domains, were more abundant in the two deep-sea species (Fig. 4a).

(a) Percentage of genes participated in different cellular processes. Each datum shows the percentage each gene group to all top 10% highly expressed genes. (b) Expression level for gene groups participated in different cellular processes. Each datum shows the percentage of average FPKM value of a gene group to average FPKM value of all top 10% highly expressed genes.
Comparison of putatively duplicated genes
There were 517, 1260 and 638 annotated duplicated gene clusters in B. pettiboneae, Lepidonotopodium sp. and H. imbricata, respectively. Their average duplicated copies were 3.6, 3.0 and 2.5 in the three species, respectively (Supplementary Table S6). GO functional annotation results of the duplicated clusters are shown in Fig. 5a. Compared with the shallow-water species, only the two deep-sea species have duplicated genes involved in cofactor binding, enzyme inhibitor and oxidoreductase activity of molecular function, and transmembrane transport of biological process. Several GO terms were only present in one deep-sea species, such as phosphatase regulator in B. pettiboneae; ribonucleoprotein complex of cellular component, carbohydrate binding, carboxylic acid binding, chromatin binding, isomerase, ligase, nucleside-triphosphatase regulator, biosynthetic process and regulation of metabolic process in Lepidonotopodium sp. The results of functional enrichment are shown in Supplementary Table S7. There were 57 GO terms significantly enriched for putatively duplicated genes in B. pettiboneae, 116 in Lepidonotopodium sp., and 37 in H. imbricata. There were 23 GO terms enriched only in the two deep-sea species (Fig. 5b), including three terminal GO terms: small molecular metabolic process, translation and DNA metabolic process. The first is related to carbohydrate and protein metabolism including pyruvate kinase and citrate synthase; and aminoacyl-tRNA synthetase and Peptidase are involved in protein metabolism. Genes enriched in translation are mainly ribosomal proteins that are responsible for protein synthesis. Genes enriched in DNA metabolism are involved in DNA recombination, integration and DNA repair.

(a) Gene Ontology (GO) distribution of putatively duplicated genes in the three species. The triangles stand for groups only present in B. pettiboneae; the squares for groups only present in Lepidonotopodium sp.; the pentagons for groups present in both deep-sea species. (b) Significantly enriched biological process GO terms of putatively duplicated genes only present in both deep-sea species (highlighted in yellow). Boxes framed in red show terminal GO terms in both deep-sea species. Data in parentheses include the number of genes associated with the listed GO term in B. pettiboneae (first number) and Lepidonotopodium sp. (second number). A dash in parenthesis indicates absence of the gene. Arrows represent the connection between two enriched GO terms at different levels. Only selected significantly enriched GO branches in deep-sea species are shown to illustrate the interspecific differences with shallow-water species.
Hemoglobins
There were 17 extracellular hemoglobin sequences from B. pettiboneae, 5 from Lepidonotopodium sp. and 7 from H. imbricata. Phylogenetic analysis showed that most of the hemoglobin sequences from the deep-sea scale worms are nested in a major clade (highlighted in red in Fig. 6). This result is consistent with the phylogenetic relationship reconstructed with concatenated sequences from the COI and 16S, 18S, and 28S rRNA genes that showed all deep-sea polynoids with a sequence in the NCBI database were derived from the same shallow-water common ancestor (Supplementary Fig. S1). There are 2 sequences from Lepidonotopodium sp. and 6 sequences from B. pettiboneae whose structures are unknown. Ten single-domain hemoglobins from 2 species of Lepidonotopodium sp., 1 species of Branchiplicatus, 2 species of Branchinologluma, and 3 species of Branchipolynoe are clustered in a clade, which is sister to a clade including all the tetra-domain hemoglobins from three species of Branchipolynoe including B. pettiboneae, B. seepensis and B. symmytilida. In each of the four domains, the B. pettiboneae sequence is sister to a clade containing the B. seepensis and B. symmytilida sequences, indicating the tetra-domain hemoglobins were present before the divergence of the Branchipolynoe spp. and the level of divergence is related to the geographic distribution of these species. These species with tetra-domain hemoglobin are in a terminal branch of the phylogenetic tree, with Branchinotogluma (a species with single-domain hemoglobin) as the sister taxon, and this result is also consistent with the phylogenetic tree based on the concatenated COI and 16S, 18S, and 28S rRNA sequences (Supplementary Fig. S1).

Sequences from Lumbricus and Glycera served as outgroups. Numbers above branches represent MP/ML bootstrap values based on 1,000 iterations, with 100 (indicated using an asterisk) as the highest value. Bootstrap values below 50 are shown only as a short dash due to the weak support. Sequences from deep-sea species are highlighted in red color.
Alignment of the single- and tetra-domain hemoglobin sequences showed that more than a quarter of amino acids (39/140) are conserved, and the first half of the sequences is more conserved than the second half (Fig. 7). Heme pocket residues, which explain the high oxygen affinity23, are conserved in tetra-domain hemoglobin sequences. There are 15 conserved amino acids in the tetra-domain sequences. Base substitution analyses of single-domain and tetra-domain hemoglobins in Branchipolynoe spp. using the branch-site model in PAML showed that three of the tetra-domains (except TD1) carry a clear signal of positive selection (Table 4). There are three sites having undergone positive selection in the tetra-domain hemoglobins (Table 4, Fig. 7).

The sequences with a blue background are tetra-domain hemoglobins from three species of Branchipolynoe. Green letters above sequences indicate the positions of the heme pocket residues in tetra-domain hemoglobin. Red letters indicate the positions of conserved amino acid residues in all hemoglobin domains. Black letters show the positions of conserved amino acids in all hemoglobins from deep-sea polynoids. Orange letters show the positions of conserved amino acids in tetra-domain hemoglobins. The asterisks show positive selected sites in tetra-domain hemoglobins. SD: single-domain; TD: tetra-domain; Bse: B. seepensis; Bsy: B. symmytilida; Bpe: B. pettiboneae; Lepw: Lepidonotopodium williamsae; Branchipc: Branchiplicatus cupreus; Branchins: Branchinotogluma segonzaci; Branchint: Branchinotogluma trifurcus.
Discussion
We provided transcriptome resources for the deep-sea scale worms Branchipolynoe pettiboneae and Lepidonotopodium sp. by high-throughput next-generation sequencing and de novo assembly. These are the only transcriptome resources for this group of scale-bearing polychaetes that are widely distributed in deep-sea chemosynthesis-based ecosystems. The resources can be used to support physiological, phylogenetic and evolutionary studies at the molecular level.
Our analysis of the codon usage and amino acid composition showed that there were differences in these two parameters between the deep-sea and shallow-water species, which is consistent with the case between deep-sea and intertidal nematodes9. In addition, there are more basic polar amino acids (arginine and histidine), and less acidic polar amino acid (aspartic acid) in deep-sea polynoids, which is consistent with a previously study showing that basic polar amino acids are more abundant in organisms living in low temperatures than in high temperatures59. The change in amino acid composition has been considered an adaptation to low temperature, which could affect protein function59.
Changes in DNA or protein sequences can be adaptive because just a few mutations can significantly affect molecular function. For example, only two amino acid substitutions in key functional positions of the skeletal actin of deep-sea fish can reduce the volume change associated with actin polymerization, which maintains the DNase I activity of actin at high pressure60. However, it is important to distinguish nonsynonymous mutation that alters amino acid from synonymous mutation that does not change amino acid. Analyzing the transcriptome-wide rates of nonsynonymous substitution to synonymous substitution can reveal genes that evolve slowly from those that evolve quickly, thereby providing a quantitative measure on the selection force15,61. In the present study, we detected 120 positively selected genes through pair-wise comparisons of homologs among the three scale worms, which can be used for more detailed studies of genetic basis of adaptation to the deep-sea environment. These genes are involved in many fundamental biological processes, including cell structure and cell adhesion, indicating reshaping of cell structures to increase toughness might have contributed to the adaption to high hydrostatic pressure in the deep sea. Positive selection in genes related to energy-related process, chromatin structure and gene expression, protein metabolism and function may have allowed the deep-sea worms to adapt to the deep sea, since high hydrostatic pressure and low temperature can inhibit gene transcription and translation, and protein function62,63. Moreover, sulfide is a major toxic substance for deep-sea organisms5. Oxidation of sulfide is a way for deep-sea animals to avoid the damage by this chemical and this process is coupled with energy production directly in the mitochondria64. Nevertheless, only protein-coding genes were included in the positive selection analysis in this study, but positive selection could occur in the regulatory regions, such as promoters and enhancers. Further analysis of positive selection in scale worms can be done when genomes of these organisms become available.
Considering that gene expression is under precise regulation in living cells, and that several genes involved in chromatin structure and regulation of gene expression exhibit signals of positive selection, we also compared the highly expressed genes among the three species of scale worms. Even though the change in environment conditions during the sampling might have affected gene expression, especially the expression of heat-shock proteins, comparison of transcriptome data should reflect most of the intrinsic gene expression difference between the deep-sea and shallow-water species. Specifically, the higher expression of hemoglobins in the deep-sea species might be a response to the hypoxic condition in the chemosynthesis-based habitats, rather than a response to higher oxygen levels they experienced during ascending. Indeed, a previous study which also involved placing the samples in a partially insulated box, indicated that ascending to surface wouldn’t eliminate the difference in expression of hemoglobin genes for the polychaete Ridgeia piscesae collected from deep-sea habitats with different DO concentrations; they also found that levels of hemoglobin gene expression of animals processed immediately were not significantly different from those processed 10–12 h after65. Our study showed that, compared with the shallow-water species, a number of conserved gene families had remarkably different expression patterns between the deep-sea and shallow-water species. In particular, in both deep-sea species, hemoglobins, a group of oxygen-binding proteins, were much more abundant in gene number (>3 times) and had much higher expression levels (>10 times), likely an adaptation to the need of using oxygen more effectively and efficiently in the deep-sea chemosynthesis habitats that often experience hypoxia1,36. In addition, the dehydrogenase gene family, which participates in carbohydrate and lipid metabolism and ATP production, has the largest number of genes in Lepidonotopodium sp. and highest expression level in B. pettiboneae. This result is consistent with the high expression of dehydrogenases in deep-sea black scabbardfish16. Furthermore, several proteins that are involved in DNA repair and recombination (i.e. reverse transcriptase, DNA breaking-rejoining domains and integrase are involved in DNA repair and recombination) were only present in the top 10% expressed genes in the two deep-sea species. The two deep-sea species also had higher abundance of proteins with zinc finger and EF hand domains which are commonly present in protein-DNA binding proteins. Together with our finding that, chromatin structure and regulation of gene expression is the largest part of positively selected genes in the two deep-sea species, these results suggest that DNA repair, recombination and integration are more active in the two deep-sea species than in the shallow-water species. This is probably related to the DNA damage caused by various stressors from the extreme deep-sea environment34.
Gene gains and losses are assumed to play a crucial role in the adaptive evolution of animals64,66. Previous studies have indicated that gene duplication may have contributed to the adaptation to cold in marine fish: there were significantly more duplicate genes in the icefish Chionodraco hamatus compared with several model fish species14, and that many of the protein-coding genes that are specifically expressed in the Antarctic fish Dissostichus mawsoni corresponded to the up-regulated gene families when compared with non-Antarctic fish based on the transcriptome data60. Even though duplicated genes might be present in shallow-water species’ genome, transcriptomic data indicates that both deep-sea species have larger numbers of duplicated genes than the shallow-water species. Different pathways were enriched for putative duplicated genes in deep-sea scale worms, especially DNA metabolism process involved in DNA recombination, DNA integration and repair. For example, there were 18 duplicated integrases in B. pettiboneae and 32 in Lepidonotopodium sp., but only 3 in H. imbricata, further supporting that DNA recombination and repair activities are more active in the two deep-sea species, consistent with a previous study that showed frequent DNA strand breakage in animals collected from deep-sea hydrothermal vents67. Furthermore, the translation pathway was significantly enriched in deep-sea species for duplicated genes. Among them are 58 and 20 duplicated ribosomal proteins in B. pettiboneae and Lepidonotopodium sp. with a total of 123 and 56 genes, respectively, indicating more ribosomes might be needed for protein synthesis in the deep-sea species.
Hemoglobins have been a research focus in deep-sea scale worms because the involvement of gene duplication as a possible adaptive mechanism to hypoxia. As oxygen-binding proteins, hemoglobins are important for energy generation. Most deep-sea scale worms are red-blooded, indicating the presence of high concentrations of hemoglobins. It has been reported that tetra-domain hemoglobins from the deep sea have higher affinity for oxygen and can store oxygen for a longer period in complete anoxia when compared with single-domain hemoglobins from their littoral relatives68,69. Tetra-domain hemoglobins were previously known only in B. seepensis and B. symmytilida23. We found 22 hemoglobin sequences in the two deep-sea scale worms, including 17 in B. pettiboneae and 5 in Lepidonotopodium sp. Consistent with their importance in oxygen storage and transport, the hemoglobins of B. pettiboneae and Lepidonotopodium sp. accounted for higher percentages of highly expressed genes and had higher expression levels than the shallow-water species H. imbricata. Branchipolynoe pettiboneae also has a tetra-domain hemoglobin, and that each of its four domains is consistent in forming the sister clade of the corresponding domain of B. seepensis and B. symmytilida. This result indicates that the formation of the tetra-domain predated the divergence of the three species of Branchipolynoe, and that B. seepensis (a Gulf of Mexico species) and B. symmytilida (an eastern Pacific species) are more closely related to each other than to B. pettiboneae (a western Pacific species), indicating that the most recent common ancestor of B. seepensis and B. symmytilida spread from western Pacific and subsequently diverged to form these two species. More taxon sampling around the world would allow for testing this hypothesis about the geographical origin of tetra-domain hemoglobin. The phylogenetic analysis showed that the three Branchipolynoe species have both single domain and tetra-domain hemoglobins. The tetra-domain hemoglobins, having high affinity to oxygen23, were only present in Branchipolynoe species, consistent with previous findings. The result from the transcriptome that the tetra-domain hemoglobin is unique in Branchipolynoe spp. should be confirmed when full genome sequences from scale worms are available. Substitution analysis of hemoglobins in Branchipolynoe spp. by branch-site model using PAML showed that all of the single domain and tetra-domain hemoglobins except TD1 were positively selected, indicating divergent evolution of hemoglobins has contributed to the adaptation in deep-sea scale worms. Specifically, the amino acid 66 C (Cysteine) in the hemoglobin is under positive selection, the same with some extracellular globins from other annelids living in sulfide-rich environments70. Cysteine in the same position was proposed to be involved in the reversible binding of sulfide in the deep-sea tube worm Riftia71. Interestingly, although Lepidonotopodium sp. does not possess tetra-domain hemoglobin, the expression level of the other type of hemoglobin was almost four times that of B. pettiboneae, indicating this scale worm has adopted a different strategy to efficiently obtain oxygen in the hypoxic deep-sea environment.
Conclusions
The first two deep-sea scale worm transcriptomes were reported in this paper. A total of 120 positively selected genes were found in pair-wise comparisons among the three species. They are involved in different fundamental biological processes, including cell structure and cell adhesion, energy-related process, chromatin structure and gene expression, signal pathway, protein metabolism and function, and transport. Among the top 10% expressed genes, globin genes in the deep-sea species were more abundant and highly expressed than in the shallow-water species; genes related to DNA repair, recombination and integration were only found in the deep-sea species. For putatively duplicated genes, GO terms related to DNA recombination and metabolism, and gene expression were only enriched in the deep-sea species. The phylogenetic analysis showed that the tetra-domain hemoglobin is unique to Branchipolynoe, and evolutionary analysis showed that three of the tetra-domains had a clear signal of positive selection. Although Lepidonotopodium sp. does not possess tetra-domain hemoglobin, its expression level of single-domain hemoglobin was four times as high as that in B. pettiboneae. The two deep-sea scale worms therefore have evolved alternative strategies in adapting to the hypoxic deep-sea environment. Taken together, our analyses revealed a number of genomic differences between the deep-sea and shallow-water scale worms. More similar comparative studies will enhance our understanding of the genomic basis of adaptation to the extreme environments in deep-sea chemosynthetic ecosystems.
Additional Information
How to cite this article: Zhang, Y. et al. Adaptation and evolution of deep-sea scale worms (Annelida: Polynoidae): insights from transcriptome comparison with a shallow-water species. Sci. Rep. 7, 46205; doi: 10.1038/srep46205 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Levin, L. A. Deep-ocean life where oxygen is scarce. Am. Sci. 90, 436–444 (2002).
Levin, L. A. et al. Hydrothermal Vents and Methane Seeps: Rethinking the Sphere of Influence. Front. Mar. Sci, 3, 72 (2016).
Watanabe, H., Fujikura, K., Kojima, S., Miyazaki, J. I. & Fujiwara, Y. Japan: vents and seeps in close proximity. In The Vent and Seep Biota (ed. Kiel, S. ) 379–401 (Springer Netherlands, 2010).
Minic, Z., Serre, V. & Hervé, G. Adaptation of organisms to extreme conditions of deep-sea hydrothermal vents. C. R. Biol. 329, 527–540 (2006).
McMullin, E. R., Bergquist, D. C. & Fisher, C. R. Metazoans in extreme environments: adaptations of hydrothermal vent and hydrocarbon seep fauna. Gravitational Space Res. 13, 13–24 (2007).
Yokoyama, S. & Tada, T. Adaptive evolution of the African and Indonesian coelacanths to deep-sea environments. Gene, 261, 35–42 (2000).
Hemmer-Hansen, J., Nielsen, E. E., Frydenberg, J. & Loeschcke, V. Adaptive divergence in a high gene flow environment: Hsc70 variation in the European flounder (Platichthys flesus L.). Heredity 99, 592–600 (2007).
Kenaley, C. P., DeVaney, S. C. & Fjeran, T. T. The complex evolutionary history of seeing red: molecular phylogeny and the evolution of an adaptive visual system in deep-sea dragonfishes (Stomiiformes: Stomiidae). Evolution 68, 996–1013 (2014).
Van Campenhout, J., Vanreusel, A., Van Belleghem, S. & Derycke, S. Transcription, signaling receptor activity, oxidative phosphorylation, and fatty acid metabolism mediate the presence of closely related species in distinct intertidal and cold-seep habitats. Genome Biol. Evol. 8, 51–69 (2016).
Bettencourt, R. et al. High-throughput sequencing and analysis of the gill tissue transcriptome from the deep-sea hydrothermal vent mussel Bathymodiolus azoricus . BMC Genomics 11, e559 (2010).
Wong, Y. H. et al. High-throughput transcriptome sequencing of the cold seep mussel Bathymodiolus platifrons . Sci. Rep. 5, e16597 (2015).
Ekblom, R. & Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 107, 1–15 (2011).
Higashi, Y., Okazaki, Y., Myouga, F., Shinozaki, K. & Saito, K. Landscape of the lipidome and transcriptome under heat stress in Arabidopsis thaliana . Sci. Rep. 5, e10533 (2015).
Coppe, A. et al. Genome evolution in the cold: Antarctic icefish muscle transcriptome reveals selective duplications increasing mitochondrial function. Genome Biol. Evol. 5, 45–60 (2013).
Yang, L., Wang, Y., Zhang, Z. & He, S. Comprehensive transcriptome analysis reveals accelerated genic evolution in a Tibet fish, Gymnodiptychus pachycheilus . Genome Biol. Evol. 7, 251–261 (2015).
Stefanni, S. et al. Transcriptome of the deep-sea black scabbardfish, Aphanopus carbo (Perciformes: Trichiuridae): tissue-specific expression patterns and candidate genes associated to depth adaptation. Int . J. Genomics 2014, e267482 (2014).
Holder, T. et al. Deep transcriptome-sequencing and proteome analysis of the hydrothermal vent annelid Alvinella pompejana identifies the CvP-bias as a robust measure of eukaryotic thermostability. Biol. Direct. 8, 1–16 (2013).
Desbruyeres, D., Gaill, F., Laubier, L. & Fouquet, Y. Polychaetous annelids from hydrothermal vent ecosystems: an ecological overview. Bull. Biol. Soc. Wash. 6, 103–116 (1985).
Rouse, G. & Pleijel, F. Palpata, Aciculata, Phyllodocida, Aphroditiformia. In Polychaetes (ed. Rouse, G. & Pleijel, F. ) 73–85 (Oxford University Press, 2001).
Norlinder, E., Nygren, A., Wiklund, H. & Pleijel, F. Phylogeny of scale worms (Aphroditiformia, Annelida), assessed from 18SrRNA, 28SrRNA, 16SrRNA, mitochondrial cytochrome c oxidase subunit I (COI), and morphology. Mol. Phylogenet. Evol. 65, 490–500 (2012).
Desbruyères, D., Segonzac, M. & Bright, M. Milestones in the discovery of hydrothermal-vent faunas. In Handbook of deep-sea hydrothermal vent fauna 2nd edn (eds Desbruyères, D. et al.) 13–25, (Biologiezentrum Linz/Austria, 2008).
Read, G. & Fauchald, K. Polynoidae Kinberg, 1856. World Polychaeta database. Available at: http://www.marinespecies.org/aphia.php?p=taxdetails&id=939 (Accessed: 7th Jan 2016).
Projecto-Garcia, J. et al. Origin and evolution of the unique tetra-domain hemoglobin from the hydrothermal vent scale worm Branchipolynoe . Mol. Biol. Evol. 27, 143–152 (2010).
Xu, T. et al. Genome-wide discovery of single nucleotide polymorphisms (SNPs) and single nucleotide variants (SNVs) in deep-sea mussels: Potential use in population genomics and cross-species application. Deep-Sea Res. Pt. II (article in press) (2016).
Feng, D. et al. Using Bathymodiolus tissue stable carbon, nitrogen and sulfur isotopes to infer biogeochemical process at a cold seep in the South China Sea. Deep-Sea Res . Pt. I 104, 52–59 (2015).
Chevaldonne, P. et al. Commensal scale worms of the genus Branchipolynoe (Polychaeta: Polynoidae) at deep-sea hydrothermal vents and cold seeps. Cah. Biol. Mar. 39, 347–350 (1998).
Pettibone, M. H. A new scale-worm commensal with deep-sea mussels on the Galapagos hydrothermal vent (Polychaeta: Polynoidae). Proc. Biol. Soc. Wash. 97, 226–239 (1984).
Pettibone, M. H. A new scale-worm commensal with deep-sea mussels in the seep-sites at the Florida Escarpment in the eastern Gulf of Mexico (Polychaeta: Polynoidae: Branchipolynoinae) Proc. Biol. Soc. Wash. 99, 444–451 (1986).
Miura, T. & Hashimoto, J. Two new branchiate scale worms (Polynoidae: Polychaeta) from the hydrothermal vent of the Okinawa through and the volcanic seamount off Chichijima Island. Proc. Biol. Soc. Wash. 104, 166–174 (1991).
Pante, E. et al. Exploration of the deep-sea fauna of Papua New Guinea. Oceanography, 25, 214–225 (2012).
Hurtado, L. A., Lutz, R. A. & Vrijenhoek, R. C. Distinct patterns of genetic differentiation among annelids of eastern Pacific hydrothermal vents. Mol. Ecol. 13, 2603–2615 (2004).
Olu, K. et al. Biogeography and potential exchanges among the Atlantic equatorial belt cold-seep faunas. PLoS One 5, e11967 (2010).
Hourdez, S. & Lallier, F. H. Adaptations to hypoxia in hydrothermal-vent and cold-seep invertebrates. Rev. Environ. Sci. Biotechnol. 6, 143–159 (2007).
Projecto-Garcia, J. et al. Selective forces acting during multi-domain protein evolution: the case of multi-domain globins. Springer Plus 4, 1–14 (2015).
Fisher, C. R. et al. Microhabitat variation in the hydrothermal vent mussel, Bathymodiolus thermophilus, at the Rose Garden vent on the Galapagos Rift. Deep-sea Res . Pt. I 35, 1769–1791 (1988).
Nakayama, N., Obata, H. & Gamo, T. Consumption of dissolved oxygen in the deep Japan Sea, giving a precise isotopic fractionation factor. Geophys. Res. Lett. 34, L20604 (2007).
Nygren, A., Norlinder, E., Panova, M. & Pleijel, F. Colour polymorphism in the polychaete Harmothoe imbricata (Linnaeus, 1767). Mar. Biol. Res. 7, 54–62 (2011).
Sun, J., Chen, Q., Lun, J. C., Xu, J. & Qiu, J. W. PcarnBase: Development of a transcriptomic database for the brain coral Platygyra carnosus . Mar. Biotechnol. 15, 244–251 (2013).
Francis, W. R. et al. A comparison across non-model animals suggests an optimal sequencing depth for de novo transcriptome assembly. BMC genomics, 14, e167 (2013).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics btu170 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 1–16 (2011).
Huang, Y., Niu, B., Gao, Y., Fu, L. & Li, W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682 (2010).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Ye, J. et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, 293–297 (2006).
Peden, J. F. Analysis of codon usage. Doctoral dissertation, University of Nottingham (2000).
Duret, L. & Mouchiroud, D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis . Proc. Natl. Acad. Sci. USA 96, 4482–4487 (1999).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Zhang, Z. et al. KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics, Proteomics & Bioinformatics 4, 259–263 (2006).
Zhang, Z. et al. ParaAT: a parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 419, 779–781 (2012).
Chen, L. Y., Zhao, S. Y., Wang, Q. F. & Moody, M. L. Transcriptome sequencing of three Ranunculus species (Ranunculaceae) reveals candidate genes in adaptation from terrestrial to aquatic habitats. Sci. Rep. 5, 10098 (2015).
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, 213–221 (2015).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, e421 (2008).
Zheng, Q. & Wang, X. J. GOEAST: a web-based software toolkit for Gene Ontology enrichment analysis. Nucleic Acids Res. 36, 358–363 (2008).
Maddison, W. P. & Maddison, D. R. Mesquite: a modular system for evolutionary analysis. Version 3.04 Available at: http://mesquiteproject.org (Accessed: 9th Nov. 2015) (2015).
Swofford, D. L. PAUP: Phylogenetic Analysis Using Parsimony, Version 4.0b10 Sinauer Associates, Inc., Sunderland (2003).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 (2011).
Yang, L. L., Tang, S. K., Huang, Y. & Zhi, X. Y. Low temperature adaptation is not the opposite process of high temperature adaptation in terms of changes in amino acid composition. Genome Biol. Evol. 7, 3426–3433 (2015).
Morita, T. Structure-based analysis of high pressure adaptation of α-actin. J. Biol. Chem. 278, 28060–28066 (2003).
Voolstra, C. R. et al. Rapid evolution of coral proteins responsible for interaction with the environment. PloS One 6, e20392 (2011).
Feller, G. & Gerday, C. Psychrophilic enzymes: hot topics in cold adaptation. Nat. Rev. Microbiol. 1, 200–208 (2003).
Simonato, F. et al. Piezophilic adaptation: a genomic point of view. J. Biotechnol. 126, 11–25 (2006).
Chen, Z. et al. Transcriptomic and genomic evolution under constant cold in Antarctic notothenioid fish. Proc. Natl. Acad. Sci. USA 105, 12944–12949 (2008).
Carney, S. L. et al. Environmental differences in hemoglobin gene expression in the hydrothermal vent tubeworm, Ridgeia piscesae . Comp. Biochem. Phys B 146, 326–337 (2007).
Ding, Y., Zhou, Q. & Wang, W. Origins of new genes and evolution of their novel functions. Annu. Rev. Ecol. Evol. Syst. 43, 345–363 (2012).
Pruski, A. M. & Dixon, D. R. Toxic vents and DNA damage: first evidence from a naturally contaminated deep-sea environment. Aquat. Toxicol. 64, 1–13 (2003).
Hourdez, S., Lallier, F. H., Martin-Jezequel, V., Weber, R. E. & Toulmond, A. Characterization and functional properties of the extracellular coelomic hemoglobins from the deep-sea, hydrothermal vent scale worm Branchipolynoe symmytilida . Proteins 34, 435–442 (1999).
Hourdez, S. & Weber, R. E. Molecular and functional adaptations in deep-sea hemoglobins. J. Inorg. Biochem. 99, 130–141 (2005).
Bailly, X. D. et al. The loss of the hemoglobin H2S-binding function in annelids from sulfide-free habitats reveals molecular adaptation driven by Darwinian positive selection. P. Natl. Acad.Sci. USA 100, 5885–5890 (2003).
Zal, F. et al. S-Sulfohemoglobin and disulfide exchange: the mechanisms of sulfide binding by Riftia pachyptila hemoglobins. P. Natl. Acad.Sci. USA 95, 8997–9002 (1998).
Acknowledgements
Feng Liu (Chinese Ocean Mineral Resources Research and Development Association) and Huaiyang Zhou (Tongji University) invited J.W.Q. to participate in a cruise to the South China Sea. Jiaolong’s pilots and operation team and the captain and crew of R/V Xiangyanghong 9 facilitated the sampling. Hiroyuki Yamamoto (JAMSTEC) invited J. S. and C. C. to join the KR15-17 cruise, the captain and crew of R/V Kairei, as well as the operation team of ROV Kaiko Mk-IV supported the sampling. Trevor Rivers (The University of Kansas) and Warren Francis (Ludwig-Maximilians-University of Munich) provided details of the sampling site for H. imbricata. This KR15-17 cruise was funded by Council for Science, Technology and Innovation (CSTI), Cross-ministral Strategic Innovation Promotion Program (SIP), “Next-generation technology for ocean resources exploration” (lead agency: JAMSTEC). This project was supported by Hong Kong Baptist University (grant no. FRG2/14-15/002 to J.W.Q.), Chinese Academy of Sciences (Strategic Priority Research Program grant no. XDB06010102 to P.Y.Q.), and an InterRidge Fellowship to J.S.
Author information
Authors and Affiliations
Contributions
J.W.Q., J.S. and P.Y.Q. conceived and coordinated the study. J.W.Q., J.S., C.C., H.K.W., and D.F. collected the deep-sea samples. Y.Z., J.S. and Yu Z. perform data analysis. Y.Z., J.W.Q. and J.M.Y.C. drafted the manuscript. All authors read, commented and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, Y., Sun, J., Chen, C. et al. Adaptation and evolution of deep-sea scale worms (Annelida: Polynoidae): insights from transcriptome comparison with a shallow-water species. Sci Rep 7, 46205 (2017). https://doi.org/10.1038/srep46205
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46205
This article is cited by
-
The first complete mitogenome of Acharax sp. (Protobranchia, Solemyida, Solemyidae): comparisons with other Solemyidae bivalves and deep-sea adaptive characteristics
Journal of Oceanology and Limnology (2023)
-
A new eyeless species of Nicon (Annelida: Nereididae) from the deep Northwest Pacific Ocean
Acta Oceanologica Sinica (2021)
-
Eyes of differing colors in Alvinocaris longirostris from deep-sea chemosynthetic ecosystems: genetic and molecular evidence of its formation mechanism
Journal of Oceanology and Limnology (2021)
-
Positive selection analysis reveals the deep-sea adaptation of a hadal sea cucumber (Paelopatides sp.) to the Mariana Trench
Journal of Oceanology and Limnology (2021)
-
Insights into high-pressure acclimation: comparative transcriptome analysis of sea cucumber Apostichopus japonicus at different hydrostatic pressure exposures
BMC Genomics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
