
Abstract
Inherited retinopathies affect approximately two and a half million people globally, yet the majority of affected patients lack clear genetic diagnoses given the diverse range of genes and mutations implicated in these conditions. We present results from a next-generation sequencing study of a large inherited retinal disease patient population, with the goal of providing clear and actionable genetic diagnoses. Targeted sequencing was performed on 539 individuals from 309 inherited retinal disease pedigrees. Causative mutations were identified in the majority (57%, 176/309) of pedigrees. We report the association of many previously unreported variants with retinal disease, as well as new disease phenotypes associated with known genes, including the first association of the SLC24A1 gene with retinitis pigmentosa. Population statistics reporting the genes most commonly implicated in retinal disease in the cohort are presented, as are some diagnostic conundrums that can arise during such studies. Inherited retinal diseases represent an exemplar group of disorders for the application of panel-based next-generation sequencing as an effective tool for detection of causative mutations.
Similar content being viewed by others


Introduction
Inherited retinal disorders (IRDs) comprise a broad spectrum of genetically heterogeneous conditions with overlapping clinical presentations. The most common diagnosis in this disease spectrum is retinitis pigmentosa (RP), with a worldwide prevalence of approximately 1/30001. RP is one of the most genetically diverse Mendelian diseases, with mutations in over 60 genes currently implicated as causative of disease, and is marked by a progressive loss of photoreceptor cells, typically involving a rod-cone degeneration with an associated loss of visual function2. Mutations in over 200 genes have been implicated in inherited retinal disorders as a whole to date3, with the result that traditional sequencing approaches are usually inadequate for the task of identifying causative mutations.
The arrival of next-generation sequencing (NGS) technologies offers for the first time an opportunity to locate these mutations. NGS technology has advanced at an exponential pace, with the cost per megabase of sequence halving regularly4,5. This has enabled enhanced diagnosis for both genetic and infectious diseases6, as well as providing insights into mutations driving tumorigenesis. Few conditions, however, have seen the same level of change as IRDs, where NGS has almost completely supplanted previous approaches.
We present results from the Target 5000 project, a NGS study aimed at identifying causative mutations in an extensive Irish IRD patient cohort. To date, probands from 309 IRD pedigrees have been sequenced. DNA samples were sequenced using a capture panel consisting of all coding exons for genes previously implicated in retinopathies as listed by Retnet3 (Supplementary Table 1). The data from prior studies have suggested that over fifty percent of disease-causing mutations will fall in the exons of known retinopathy genes7, and hence targeting these exons allows rapid and cost-effective identification of causative mutations for the majority of patients, with improved sensitivity for mutations in the target region compared to whole-exome capture8.
Employing target-panel capture and sequencing and ensemble prediction methods for novel variant classification, candidate mutations were identified in 57% of pedigrees, despite the substantial genetic heterogeneity of these conditions. Many novel and previously reported mutations were identified in this IRD cohort, as were new ocular disease phenotypes associated with genes previously implicated in other retinal disorders.
Results
Sample Acquisition and Sequencing
Probands from a total of 309 Irish IRD pedigrees were sequenced. Following informed consent, samples were taken and clinical examinations performed. Patients over 18 years with an IRD were eligible to enrol in the study. A full breakdown of the frequency of different conditions is provided in Fig. 1.

Breakdown of patient cohort by condition.
DNA was isolated and samples were prepared, target-captured and sequenced as described in the Methods section. The list of genes sequenced is given as Supplementary Table 1. Overall sequence quality was high. An average of 98.9% of the targeted coding sequence in each sample had 8X coverage or better (interquartile range 98.6–99.5%), with 95% covered at 30X coverage or better.
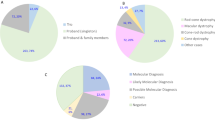
Overall, causative mutation(s) could be identified in 57% (176/309) of sequenced pedigrees, although this proportion differed substantially between different conditions (see Fig. 2). A substantial number of novel mutations that had not previously been associated with retinal disease were identified (Table 1).

Prevalence of causative mutations in specific genes in patients with an initial diagnosis of:
Top-left: Retinitis pigmentosa. Top-right: Usher Syndrome. Bottom-left: Stargardt Disease or macular dystrophy. Bottom-right: Leber Congenital Amaurosis or Early-Onset Severe Retinal Degeneration.
Stargardt Disease
The most commonly implicated gene in Stargardt disease was ABCA4, causing recessive Stargardt disease or Fundus Flavimaculatus in 32 pedigrees. Although Stargardt disease is not the most common condition in the Irish IRD patient cohort, it is very genetically homogeneous: 73% of sequenced Irish Stargardt pedigrees showed causative mutations in ABCA4, with many of the remainder having one identifiable ABCA4 mutation but not two. ABCA4 mutations are the primary cause of Stargardt disease and autosomal-recessive cone-rod dystrophy, in addition to causing a small number of cases of RP9; ABCA4 was therefore the single most commonly implicated gene across all conditions in this study. This cohort included one pedigree with a rare case of dominant Stargardt-like disease caused by a mutation in the PROM1 gene, (NM_006017.2:c.1117C > T,p.Arg373Cys), which has previously been identified as causative of dominant maculopathy10. In addition, mutations in BEST1 were identified in one pedigree in the cohort, leading to a rediagnosis of Best Vitelliform Macular Dystrophy.
Leber Congenital Amaurosis (LCA) and Early-Onset Severe Retinal Degeneration (EOSRD)
The cohort of IRD patients with LCA/EOSRD was relatively small, comprising 18 patients across 11 pedigrees. The mutation NM_001122769.2:c.1756A > T,p.Lys586* in the LCA5 gene, which encodes the Lebercillin protein, segregated with the disease in a recessive LCA pedigree with two affected members. This mutation has not previously been reported as disease causing, although the variant was reported by the Exome Aggregation Consortium (ExAC)11 at an allele frequency of 1/121,012. No homozygotes have previously been reported. As a premature stop mutation in a gene implicated in LCA and showing segregation with disease, it is highly likely to be the causative mutation. The homozygous mutation (NM_000554.4:c.206G > A,p.Arg69His) in the CRX gene was observed in a simplex pedigree with no other candidate mutations. The Arg69His substitution has not been implicated in disease before, but has been observed in ExAC at extremely low allele frequency (2/121,402), with no homozygotes reported. The mutation is predicted by the ensemble model to cause disease and is located in the crucial homeodomain of CRX, a domain previously reported to be sensitive to missense mutations12. Sanger sequencing of the patient’s mother, who retains normal visual function, confirmed the heterozygous presence of the mutation, indicating that the mode of inheritance is indeed recessive.
In one interesting simplex case compound heterozygous frameshifts in the NRL gene, (NM_006177.3:c.16delA,p.Ser6fs) and (NM_006177.3:c.386delC,p.Ala129fs), were found in a patient with atypical LCA marked by unusually good retention of visual acuity despite the presence of typical LCA features such as congenital nystagmus, extinguished ERG and onset of visual symptoms at a very young age. Dominant-acting NRL mutations have been reported in the past13, and so molecular cloning of the entire region was used to verify that the two mutations were on separate chromosomes and did not merely represent a single haplotype. Mutations in NRL have also been implicated previously in recessive inherited retinal disease14, however we have been unable to find another reported case of a patient with two frameshift or nonsense mutations in NRL. As such, this is a phenotype of interest, as the first homozygous NRL-null case reported in humans.
At 46 years the proband retained best-corrected Snellen visual acuity of 6/60 and 6/15 in the right and left eyes respectively. Visual fields were concentrically constricted to within 10° of fixation to the Goldmann IV4e target. No convincing rod or cone full-field ERG responses were recordable, although delayed and reduced amplitude cone responses were recordable 10 years previously. Fundoscopy at 46 years of age revealed extensive retinal and choroidal atrophy peripherally, with better preservation at the posterior pole in each eye. Scattered clumped pigment deposits were observed, consistent with existing observations of patients with recessive NRL-based disease14.
Retinitis Pigmentosa (RP)
The largest disease cohort comprised patients with RP, accounting for 37.9% of the total patient cohort. The most commonly implicated genes for RP varied based on inheritance: for dominant RP, the most commonly implicated genes were RHO (8.6%), RP1 (8.6%) and PRPH2 (3.8%). This may underestimate the true prevalence of RHO mutations in autosomal dominant RP, as pre-NGS sequencing studies had been performed in the Irish IRD population focusing solely on exons of RHO15,16. As a result, a number of patients with RHO mutations had already been identified, and this subpopulation of Irish IRD patients did not participate in the current study, resulting in a likely underestimate of the frequency of RHO-linked adRP.
The primary contributor to the high prevalence of RP1 mutations in dominant RP was a frameshift mutation in RP1 (NM_006269.1:c.2285_2289delTAAAT,p.Leu762fs) that was observed to segregate with disease in five dominant RP pedigrees, including one large pedigree with four affected members and several unaffected members, and was never observed in pedigrees with other conditions. Although these patients presented to the clinic as unrelated probands and were not geographically clustered, it is anticipated that they likely share a common ancestor and form a contiguous super-pedigree. This RP1 mutation has not been previously reported in dbSNP or ExAC, nor was it observed in a recent study of RP patients in Northern Ireland17 but is the most frequent single mutation causing RP in the current study.
Similarly, the primary contributor to the high prevalence of PRPH2 mutations is the S212G (NM_000322.4:c.634A > G,p.Ser212Gly) mutation which has previously been identified as causative of disease in the Irish population18. As with the RP1 p.Leu762fs mutation, patients carrying the S212G mutation likely share a common ancestor, although they presented as unrelated probands in the clinic.
In cases of X-linked RP, nearly half of all sequenced pedigrees (10/21) showed mutations in the RPGR gene, representing almost all of the XLRP pedigrees for which mutations could be identified in this study, with the exception of two pedigrees with RP2 mutations. The RPGR mutations included the previously-reported G436D (NM_001034853.1:c.1307G > A,p.Gly436Asp) mutation. However, most mutations in RPGR were premature stop and frameshift mutations. Several of these were novel mutations that have neither been reported previously nor implicated in disease (Table 1). There was also one novel amino acid substitution in the RPGR gene (p.Thr99Ala) that segregated with disease in a small pedigree. This region of the protein is highly conserved and quite sensitive to mutation19; a different mutation in the same codon (p.Thr99Asn)19, as well as a mutation in the adjacent codon (p.His98Gln)20 have been implicated in disease. Simulated folding of both the native and the novel p.Thr99Ala proteins using the I-TASSER suite21 showed a disruption in the beta-propeller domain that contains residue Thr99, converting that domain from beta-sheet to coil conformation. Such a conformational change would be likely to severely affect protein function (Fig. 3).

Left: A simulated fold of the beta-propeller domain in the native RPGR sequence shows the expected seven beta sheet propellers forming correctly. Thr99 is highlighted in green. Right: A simulated fold of the same domain in RPGR-T99A. Ala99 is highlighted in green. The entire propeller containing residue 99 has converted from sheet to coil conformation.
For recessive and simplex forms of RP, the results obtained were more disparate. Notably, however, four pedigrees within the simplex/recessive RP cohort were found to carry the BBS1 mutation NM_024649.4:c.1169T > G,p.Met390Arg, which has been associated with Bardet-Biedl syndrome. In one of these cases, the patient was noted to have mild mental retardation consistent with Bardet-Biedl syndrome. However, the other three patients showed none of the extraretinal features of the disease. These cases therefore continued to be classified as nonsyndromic RP. This is consistent with reports from other groups, which have identified that BBS1 p.Met390Arg can cause either Bardet-Biedl syndrome or nonsyndromic RP in different pedigrees22.
Two cases in the recessive/simplex RP cohort were particularly noteworthy: A homozygous premature stop mutation in the GNAT1 gene (NM_000172.3:c.904C > T,p.Gln302*) in a case of simplex RP, which has been reported in an earlier paper23 and a homozygous frameshift in the SLC24A1 gene (NM_004727.2:c.2679delT,p.Asn893fs) in a pedigree with two affected individuals. In both situations, the mutation segregated with the retinopathy (although in the case of the GNAT1 mutation, there was only one patient who carried it homozygously and only this individual was affected in the pedigree). Both of these genes have been implicated in congenital stationary night-blindness (CSNB), but neither gene has previously been implicated in RP. Although both CSNB and RP affect rod photoreceptor cells, rod cells do not die in significant numbers in CSNB, with the result that the disease is largely stationary and does not affect central or colour vision, although dark vision is entirely lost24. This finding therefore represents a novel clinical phenotype caused by SLC24A1 mutations.
In both the GNAT1 and the SLC24A1 pedigrees, the mutations were recessive premature-stop mutations, causing loss of protein function. In both pedigrees onset of the disease was late for RP, with a mild, slowly-progressing course. Similar to the GNAT1 phenotype23, patients in the SLC24A1 pedigree presented with lifelong night-blindness, but with very distinctive, albeit mild, symptoms of RP including progressive mid-peripheral visual field loss, early appearance of cataracts and unmistakable pigmentary retinopathy on fundus photography (Fig. 4).

Fundus photograph of an affected patient from the SLC24A1-RP pedigree.
Image quality is suboptimal because of the presence of cataracts, although pigmentary deposits, supporting the diagnosis of retinitis pigmentosa, are clearly visible.
Notably, no other plausibly causative mutations were observed in either pedigree in the retinal genes included in the study. The data suggest that in both cases, a severe, homozygous mutation in a known CSNB disease gene has caused a mild, late-onset form of RP, and we therefore hypothesize that this may represent a general pattern in retinal disease. Additional observations of the phenotype resulting from severe mutations in CSNB-associated genes in other pedigrees will be required to confirm or refute this.
Usher Syndrome
A total of 32 pedigrees with Usher syndrome were sequenced during the study, with type II disease being the most common (23 pedigrees), followed by type I (5 pedigrees). 4 other pedigrees had atypical or type III Usher syndrome. Consistent with previous work25,26, Usher Type I pedigrees were found to be predominantly (80%) caused by MYO7A mutations, while Usher Type II pedigrees were most commonly (74%) caused by mutations in USH2A. No causative mutations could be found in patients with atypical Usher syndrome, although two mutations were identified in the CLRN1 gene in a patient with type III Usher syndrome.
Choroideremia
Choroideremia is caused only by mutations in the CHM gene. As a result, detection was relatively uncomplicated and efficiencies were good. A total of 12 pedigrees with choroideremia were included in the study. Detection rates were the highest of all conditions included in this study; only two of the twelve pedigrees could not be solved. Three pedigrees in this cohort are particularly noteworthy: In one, the mutation NM_000390.2:c.1376C > G,p.Thr459Arg in the CHM gene was observed in all three affected members and no unaffected members, with no other relevant mutations being detected in that gene. This mutation is predicted by Human Splicing Finder27 to affect correct splicing and has not previously been reported as causative, and so this finding is of clinical relevance for future choroideremia diagnoses. In a second pedigree, a novel 6.3 kb deletion (hg19 co-ordinates chrX:85233437-85239772) was found to remove exons 3 and 4 of the CHM gene.
A third pedigree, however, was an unusual case. NGS of the proband revealed a deleterious mutation in the CHM gene: NM_000390.2 c.715C > T,p.Arg239*, which was confirmed by Sanger sequencing. The pedigree structure was consistent with X-linked inheritance, with no affected females. A second affected member of the pedigree was Sanger sequenced, but was found not to carry the CHM mutation. At the time of assessment, this patient had an end-stage retinopathy which could not be cleanly classified, although it was presumed before sequencing was performed that they carried the same mutation and condition as the rest of the pedigree (see Fig. 5). The absence of the mutation was confirmed again using a fresh sample from the patient, indicating that the CHM mutation was definitively not present. NGS of this patient revealed a homozygous mutation in the RPE65 gene (NM_000329.2:c.271C > T,p.Arg91Trp), which is known to be associated with retinitis pigmentosa28. Subsequent clinical work confirmed a diagnosis of RPE65-RP, making this was a very unusual case of two rare, genetically distinct Mendelian retinal diseases segregating within the same pedigree.

Left: Fundus photograph of a patient from the RPE65/CHM pedigree carrying a CHM mutation shows a classic choroideremia phenotype of optic disc pallor, mild vascular attenuation and relative preservation of the macula. Right: Fundus photograph of the patient from the RPE65/CHM pedigree carrying the homozygous RPE65 mutation shows some differences, including some bone-spicule deposits and poorer preservation of the macula.
Discussion
Adopting a target panel-based NGS approach targeting the exons of known retinal disease genes, causative mutations could be identified in 57% of pedigrees (a breakdown by condition is given in Fig. 2). The fraction of pedigrees for which causative mutations could be identified varied widely between retinal conditions, with conditions such as Stargardt disease and choroideremia having the highest detection rates (70–86%).
A major novel finding from this study was the association of two genes, GNAT1 and SLC24A1, with RP that had previously only been associated with congenital stationary night blindness (CSNB). Although both retinitis pigmentosa and stationary night blindness affect rod photoreceptor cells, they are considered to be distinct conditions. In both cases, the mutation was a complete loss of function, either a homozygous premature stop or a homozygous frameshift. This suggests the intriguing possibility of a recurring pattern: Severe variants in CSNB-associated genes can cause a mild form of RP, potentially blurring the distinction between CSNB- and RP -associated genes, although the data is too limited for strong conclusions to be drawn as yet.
The results of this study emphasize the diversity of mutations underlying IRDs in the Irish population and the significant value of targeted NGS for IRDs. More than forty novel, previously unreported mutations were identified in this patient cohort (Table 1). We demonstrate the application of novel ensemble prediction methods and protein folding simulations for variant effect prediction (Fig. 3). In other cases, mutations in unexpected genes, such as BBS1 mutations in cases of nonsyndromic RP, underscored the significant overlap between different conditions in terms of clinical presentations, as well as the difficulty in identifying the causative gene based on clinical examination alone. The observation of one pedigree in which two IRDs are segregating (RPE65-RP and CHM-Choroideremia) serves to emphasise the essential role that NGS will play in the future diagnosis of this genetically heterogeneous group of conditions.
The pedigrees for which a mutation could not be identified are likely to be a mixture of those which could not be observed, such as mutations in promoter regions and other non-coding regions or genes not included in the panel, and those where the true mutations were in the sequenced region but could not be uniquely identified as causative because of the presence of multiple candidates. It is difficult to assess what proportion of pedigrees fall into each category, however an estimate can be made given that just over 25% of pedigrees had no candidate variants remaining in relevant genes after common and synonymous mutations were filtered out of the analysis. We suggest that this may be a reasonable, albeit conservative, lower bound for the fraction of pedigrees that may have non-coding or novel-gene mutations requiring whole-exome or whole-genome sequencing.
This issue informs the question of whether to select whole-genome or targeted-panel sequencing for future studies. Costs of NGS can be broken down between library preparation and sequencing. Library preparation for a targeted panel of genes is more expensive in terms of time and reagents than preparing a whole-genome library, however, this is outweighed by the cost of deep-coverage whole-genome sequencing, which is still about $1000/sample, 10 or 20 times the cost for sequencing of pooled target-capture samples.
Until whole-genome sequencing costs fall to a half or a quarter of current levels, therefore, there is still a role for targeted sequencing due to lower cost and the simplicity of analysis. For the near-term future, we recommend a two-tier approach to genetic analysis of IRDs: targeted sequencing of a core panel of exons to identify mutations in known genes, followed by whole-exome or whole-genome sequencing for pedigrees in which a mutation could not be found by the targeted approach. This applies even when the aim of the study is to identify non-coding mutations and new disease genes, as the cost savings from eliminating 50–60% of patients with coding mutations in known genes from the larger-scale sequencing study would more than outweigh the cost of performing targeted sequencing. The combination of these approaches, in concert with the increasing range of known retinopathy genes and the decreasing cost of wider-scale sequencing approaches, is likely to improve mutation detection rates even further in the next few years.
Methods
Patient Identification and Recruitment
Probands and other family members were primarily assessed at the Research Foundation of the Royal Victoria Eye and Ear Hospital (Dublin, Ireland) and the Mater Misericordiae University Hospital (Dublin, Ireland). With informed consent, best-corrected visual acuity was assessed using revised 2000 ETDRS charts (Precision Vision, La Salle Il, USA). Color vision was examined using the Lanthony desaturated panel D-1529 under standardised lighting conditions. Goldmann perimetry was used to assess the peripheral visual fields to the IV4e, I4e and 04e targets. Full-field electroretinograms were performed according to ISCEV standards30 using a Roland Consult RETI-port retiscan (Brandenburg an der Havel, Germany). Fundus color and autofluorescence photography was performed using a Topcon CRC50DX. Spectral domain optical coherence tomography was performed using a Cirrus HD-OCT (Carl Zeiss Meditec, Germany).
DNA Isolation and Sequencing
Following informed consent, blood samples were collected from patients after clinical assessment. DNA was isolated from 2 ml of patient blood and fragmented for sequencing by ultrasonication in a Diagenode Bioruptor (Diagenode s.a., Belgium) to an average fragment size of 200–250 bp.
Sequencing libraries were generated and target capture was performed initially using the Agilent Sureselect XT2 kit (Agilent Technologies, Santa Clara, CA). Later captures used a redesigned panel with the Roche Nimblegen SeqCap EZ kit (Roche), incorporating new genes implicated in retinopathies since the design of the earlier panel. For both kits, captures were performed according to the manufacturer’s recommendations. Exons for all genes previously implicated in retinal degeneration, as listed by Retnet at the time of capture panel design, were included as capture targets, plus 100 bp surrounding the CEP290 intronic mutation implicated in LCA31. Additional intronic regions in ABCA432 and USH2A33 that are commonly implicated in disease were not included in the panel but were sequenced by single-read sequencing in relevant patients when causative mutations were not detected by targeted sequencing. These regions will be included in future versions of the panel. In the earlier capture design, UTRs were also included, but these were excluded in later panels to improve exon coverage and sequencing throughput. The total size of the captured region was 1,490 kb for the earlier captures and 728 kb for the later captures.
Captured patient DNA was multiplexed into either 24- or 96-sample pools and sent for sequencing. Sequencing of 96-sample pools was performed off-site by BGI Tech using an Illumina HiSeq 2000 (Illumina, San Diego, CA). 24-sample pools were sequenced locally using an Illumina MiSeq. Confirmatory single-read sequencing was also performed to verify the presence of candidate mutations.
Data Analysis
Sequence data were demultiplexed and mapped to the human genome (hg19) using BWA version 0.7.1234. Duplicate reads were flagged using Picard version 1.10635 and downstream analysis and variant calling were performed using GATK version 3.3.036 according to the protocol specified in the GATK Best Practices Workflow, with the notable difference that hard-filtering rather than variant quality score recalibration (VQSR) was used to filter variants, due to the small size of the capture area. Variants filtered by this method were not discarded, but instead marked as potential sequencing artefacts.
The list of identified variants was annotated with snpEFF37 and dbNSFP38, and an ensemble model was used for classification of novel variants. Commonly used variant effect prediction software is based on machine learning approaches that learn a classifier model based on patterns in datasets of known pathogenic and neutral mutations39,40. In the fields of statistics and machine learning, ‘ensemble’ classifier models are commonly used which learn meta-classifiers using the predictions of existing individual classifier models as inputs. Such models frequently have performances superior to any individual model in the ensemble41, particularly if, as is the case with variant effect prediction, the individual models employ different approaches, features and training sets to generate their models and therefore have errors that should be substantially decorrelated with each other. The primary tool adopted for novel variant pathogenicity prediction was therefore an ensemble prediction model42, which shows significant improvements in accuracy compared to many tools, but which to our knowledge has not yet been used in studies of IRDs.
Synonymous variants and common polymorphisms were filtered out, and the remaining list of rare variants with the potential to affect protein sequence was output for manual curation. The output for each patient also included a list of coding regions where coverage was insufficient for reliable variant calling.
Ethical Approval
Ethical approval for this study was granted by the ethics committee of the Royal Victoria Eye and Ear Hospital prior to commencement of this study. All work was carried out in accordance with the approved guidelines. All patients gave written informed consent before recruitment to the study. No patients under 18 years of age were included in the study.
Additional Information
How to cite this article: Carrigan, M. et al. Panel-Based Population Next-Generation Sequencing for Inherited Retinal Degenerations. Sci. Rep. 6, 33248; doi: 10.1038/srep33248 (2016).
References
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet 368, 1795–1809 (2006).
Daiger, S. P., Sullivan L. S. & Bowne S. J. Genes and mutations causing retinitis pigmentosa. Clin Genet. 84(2), 132–134 (2013).
RetNet. Available at: http://www.sph.uth.tmc.edu/RetNet/ (Accessed Oct 21, 2015).
Gullapalli, R. R., Desai, K. V., Santana-Santos, L., Kant, J. A. & Becich, M. J. Next generation sequencing in clinical medicine: Challenges and lessons for pathology and biomedical informatics. J Pathol Inform 3, 40 (2012).
Lecuit, M. & Eloit, M. The potential of whole genome NGS for infectious disease diagnosis. Expert Rev. Mol. Diagn. 15, 1517–1519 (2015).
Naccache, S. N. et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 24, 1180–1192 (2014).
Siemiatkowska, A. M., Collin, R. W. J., den Hollander, A. I. & Cremers, F. P. M. Genomic approaches for the discovery of genes mutated in inherited retinal degeneration. Cold Spring Harb Perspect Med 4, (2014).
Consugar, M. B. et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet. Med. 17, 253–261 (2015).
Maugeri, A. et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am. J. Hum. Genet. 67, 960–966 (2000).
Michaelides, M. et al. The PROM1 mutation p.R373C causes an autosomal dominant bull’s eye maculopathy associated with rod, rod-cone, and macular dystrophy. Invest. Ophthalmol. Vis. Sci. 51, 4771–4780 (2010).
Exome Aggregation Consortium (ExAC), Cambridge, MA. Available at: http://exac.broadinstitute.org (Accessed Oct 21, 2015).
Bessant, D. A. et al. A mutation in NRL is associated with autosomal dominant retinitis pigmentosa. Nat. Genet. 21, 355–356 (1999).
Chen, S. et al. Functional analysis of cone-rod homeobox (CRX) mutations associated with retinal dystrophy. Hum. Mol. Genet. 11, 873–884 (2002).
Nishiguchi, K. M. et al. Recessive NRL mutations in patients with clumped pigmentary retinal degeneration and relative preservation of blue cone function. Proc. Natl. Acad. Sci. USA. 101, 17819–17824 (2004).
Farrar, G. J. et al. Autosomal dominant retinitis pigmentosa: a mutation in codon 178 of the rhodopsin gene in two families of Celtic origin. Genomics 11, 1170–1171 (1991).
al-Jandal, N. et al. A novel mutation within the rhodopsin gene (Thr-94-Ile) causing autosomal dominant congenital stationary night blindness. Hum. Mutat. 13, 75–81 (1999).
Zhao, L. et al. Next-generation sequencing-based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Hum. Genet. 134, 217–230 (2015).
Farrar, G. J. et al. Autosomal dominant retinitis pigmentosa: a novel mutation at the peripherin/RDS locus in the original 6p-linked pedigree. Genomics 14, 805–807 (1992).
Ghosh, A. K. et al. Human retinopathy-associated ciliary protein retinitis pigmentosa GTPase regulator mediates cilia-dependent vertebrate development. Hum. Mol. Genet. 19, 90–98 (2010).
Meindl, A. et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat. Genet. 13, 35–42 (1996).
Yang, J. et al. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015).
Wang, F. et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum. Genet. 133, 331–345 (2014).
Carrigan, M. et al. A novel homozygous truncating GNAT1 mutation implicated in retinal degeneration. Br J Ophthalmol, doi: 10.1136/bjophthalmol-2015-306939 (2015).
Zeitz, C., Robson, A. G. & Audo, I. Congenital stationary night blindness: an analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog Retin Eye Res 45, 58–110 (2015).
Jaijo, T. et al. MYO7A mutation screening in Usher syndrome type I patients from diverse origins. J. Med. Genet. 44, e71 (2007).
Bessant, D. A., Payne, A. M., Plant, C., Bird, A. C. & Bhattacharya, S. S. Further refinement of the Usher 2A locus at 1q41. J. Med. Genet. 35, 773–774 (1998).
Desmet, F.-O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67 (2009).
Samardzija, M. et al. R91W mutation in Rpe65 leads to milder early-onset retinal dystrophy due to the generation of low levels of 11-cis-retinal. Hum. Mol. Genet. 17, 281–292 (2008).
Lanthony, P. The desaturated panel D-15. Doc Ophthalmol 46, 185–189 (1978).
Marmor, M. F. et al. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol 118, 69–77 (2009).
den Hollander, A. I. et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 79, 556–561 (2006).
Bauwens, M. et al. An augmented ABCA4 screen targeting noncoding regions reveals a deep intronic founder variant in Belgian Stargardt patients. Hum. Mutat. 36, 39–42 (2015).
Steele-Stallard, H. B. et al. Screening for duplications, deletions and a common intronic mutation detects 35% of second mutations in patients with USH2A monoallelic mutations on Sanger sequencing. Orphanet J Rare Dis 8, 122 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Picard. Available at: http://broadinstitute.github.io/picard/
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Liu, X., Wu, C., Li, C. & Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Non-synonymous and Splice Site SNVs. Hum. Mutat., doi: 10.1002/humu.22932 (2015).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter 7, Unit7.20 (2013).
Shihab, H. A. et al. Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum. Genomics 8, 11 (2014).
Polikar, R. Ensemble based systems in decision making. IEEE Circuits and Systems Magazine 6, 21–45 (2006).
Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 24, 2125–2137 (2015).
Acknowledgements
This research was supported by grant awards from Fighting Blindness Ireland (FB Irl), The Health Research Board of Ireland (HRB), The Medical Research Charities Group (MRCG), the Irish Research Council (IRC) and Science Foundation Ireland (SFI). We would also like to acknowledge the assistance of Hugh Nolan at the Royal Victoria Eye & Ear Hospital, Dublin, Adrian Dockery at Trinity College Dublin and Elaine Kenny at the Trinity Genome Sequencing Laboratory, Dublin.
Author information
Authors and Affiliations
Contributions
M.C. was the primary author of the manuscript and responsible for sample preparation, bioinformatics, data analysis and generation of Figures. E.D., C.M., K.S., T.S., D.K. and P.K. were responsible for clinical assessment of patients and recruitment to the study, in addition to sample collection. CMcD was responsible for some data analysis and confirmatory sequencing in several key pedigrees. A.G., D.K., P.H., P.K. and G.J.F. were responsible for study design and oversight. G.J.F. was the PI of the lab in which sequence analysis was carried out.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Carrigan, M., Duignan, E., Malone, C. et al. Panel-Based Population Next-Generation Sequencing for Inherited Retinal Degenerations. Sci Rep 6, 33248 (2016). https://doi.org/10.1038/srep33248
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep33248
This article is cited by
-
Detailed analysis of an enriched deep intronic ABCA4 variant in Irish Stargardt disease patients
Scientific Reports (2023)
-
Clinical exome sequencing for inherited retinal degenerations at a tertiary care center
Scientific Reports (2022)
-
Genetic characteristics and epidemiology of inherited retinal degeneration in Taiwan
npj Genomic Medicine (2021)
-
Target 5000: a standardized all-Ireland pathway for the diagnosis and management of inherited retinal degenerations
Orphanet Journal of Rare Diseases (2021)
-
A FBN1 variant manifesting as non-syndromic ectopia lentis with retinal detachment: clinical and genetic characteristics
Eye (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
