
Abstract
We aimed to assess the impact of L-carnitine on plasma Lp(a) concentrations through systematic review and meta-analysis of available RCTs. The literature search included selected databases up to 31st January 2015. Meta-analysis was performed using fixed-effects or random-effect model according to I2 statistic. Effect sizes were expressed as weighted mean difference (WMD) and 95% confidence interval (CI). The meta-analysis showed a significant reduction of Lp(a) levels following L-carnitine supplementation (WMD: −8.82 mg/dL, 95% CI: −10.09, −7.55, p < 0.001). When the studies were categorized according to the route of administration, a significant reduction in plasma Lp(a) concentration was observed with oral (WMD: −9.00 mg/dL, 95% CI: −10.29, −7.72, p < 0.001) but not intravenous L-carnitine (WMD: −2.91 mg/dL, 95% CI: −10.22, 4.41, p = 0.436). The results of the meta-regression analysis showed that the pooled estimate is independent of L-carnitine dose (slope: −0.30; 95% CI: −4.19, 3.59; p = 0.878) and duration of therapy (slope: 0.18; 95% CI: −0.22, 0.59; p = 0.374). In conclusion, the meta-analysis suggests a significant Lp(a) lowering by oral L-carnitine supplementation. Taking into account the limited number of available Lp(a)-targeted drugs, L-carnitine might be an effective alternative to effectively reduce Lp(a). Prospective outcome trials will be required to fully elucidate the clinical value and safety of oral L-carnitine supplementation.
Similar content being viewed by others

Introduction
In 1963, Kåre Berg discovered lipoprotein(a) [Lp(a)] in human plasma, a low-density lipoprotein (LDL)-like lipoprotein particle with atherogenic and thrombotic properties1. Lp(a) is composed by a central LDL-like lipoprotein core particle with apolipoprotein B (apoB) covalently bound to glycoprotein apo(a), resulting in differing structure, physical and chemical properties compared with LDL1,2. The levels of Lp(a) in human plasma are genetically modulated and Precocious Coronary Artery Disease (PROCARDIS) study showed that two Lp(a) single nucleotide polymorphisms (SNPs) accounted for 36% of variation in Lp(a) levels3. The levels of Lp(a) vary among populations; African descendants have increased Lp(a) levels as compared to Caucasians and Asian descendants4. However, different conditions such as diabetes, endocrine disorders, renal and liver failure or the acute-phase response might influence the synthesis or catabolism of Lp(a)5,6. The development of immunoassays to precisely measure Lp(a) levels in plasma was impeded by the structural complexity and size heterogeneity of Lp(a)3.
Many studies have shown that increased plasma Lp(a) levels are associated with coronary artery disease7,8, peripheral arterial disease9, cerebrovascular disease10, abdominal aortic aneurysm, aortic valve stenosis and calcification11 and venous thromboembolism12. Moreover, elevated Lp(a) levels have been found to be associated with increased risks for major adverse cardiac events (MACE)13. The atherogenic impact influenced by the effective binding of oxidized phospholipids by Lp(a), the expression of adhesion molecules by Lp(a)14, the deposit of Lp(a) in the arterial wall and the prothrombotic effect caused by the homology of apo(a) to plasminogen are potential mechanisms by which Lp(a) raises cardiovascular (CV) risk15,16. After migration from plasma into the arterial intima, Lp(a) joins to the extracellular matrix through both apo(a) and apoB components, resulting in the growing of the atherosclerotic plaque17.
In 2010, the European Atherosclerosis Society (EAS) Consensus Panel recommended a target level for Lp(a) < 50 mg/dL, as a feature of global CV risk18. The treatment of choice for the patients with increased Lp(a) levels is niacin or nicotinic acid (1–3 g/day), as recommended by the European Society of Atherosclerosis (EAS) and American Heart Association (AHA)/American Stroke Association (ASA) guidelines18,19. However, after negative trials with niacin, statins are worldwide recommended to reduce the global CV risk20. Moreover, observational studies suggested that the enhanced CV risk associated with increased Lp(a) levels is no longer observed when LDL-C levels are treated to aggressive levels, suggesting intensification of statin therapy and LDL-C lowering as a potential option in subjects with Lp(a) elevation13,20. Niacin therapy produces side-effects via stimulation of prostaglandin D(2) and E(2) synthesis, posing another obstacle to its use for Lp(a) elevation in clinical practice21. Therefore, a new therapy to target high Lp(a) levels is considered an important and unmet clinical need22. Besides niacin and intensive statin therapy, tibolone has also been investigated in patients with elevated Lp(a) levels23. Other drugs currently under development include cholesteryl ester transfer protein (CETP) inhibitors24, proprotein convertase subtilisin/kexin type-9 (PCSK9) inhibitory monoclonal antibodies24,25, mipomersen, an antisense oligonucleotide (ASO) against apoB mRNA26, eprotirome, a thyroid analogue27, lomitapide, a microsomal triglyceride transfer protein inhibitor28 and ASO therapy (ISIS APO(a) Rx) directly against apo(a)29,30. In addition, there has been a surge of interest to screen natural products (nutraceuticals/functional foods) for their effects on Lp(a) concentration31,32.
Different studies have indicated that L-carnitine, an amino acid involved in mitochondrial fatty acid oxidation and ATP production, might be associated with reduction in Lp(a) levels33. The metabolism of L-carnitine is complex and involves formation of many short and long chain acylated forms, as well as metabolism to its core structure31,32,33. In addition, there are substantial alterations in the metabolism of L-carnitine depending upon whether it is provided as intravenous supplement, such as during hemodialysis, or as oral supplement34. With the latter route of supplementation, gut microbes have been shown to significantly impact L-carnitine metabolism and to convert it into alternative metabolites, linked to increased atherosclerosis risk35,36.
Therefore, we aimed to assess the impact of L-carnitine supplementation and type of administration on plasma Lp(a) concentrations through systematic review of literature and meta-analysis of available randomized controlled trials (RCTs).
Methods
Search Strategy
This study was designed according to the guidelines of the 2009 preferred reporting items for systematic reviews and meta-analysis (PRISMA) statement37. SCOPUS (http://www.scopus.com) and Medline (http://www.ncbi.nlm.nih.gov/pubmed) databases were searched and the search was limited to the randomized controlled studies (RCTs) carried out up to January 31, 2015 investigating the potential effects of L-carnitine supplementation on Lp(a) concentrations. The databases were searched using the following search terms in titles and abstracts (also in combination with MESH terms): (L-carnitine) AND Lp(a), or (L-carnitine) AND Lipoprotein(a). The wild-card term “*” was used to increase the sensitivity of the search strategy. No language restriction was used in the literature search. The search was limited to studies in human. Selected articles were searched to identify further relevant studies. Two reviewers (CS and AS) evaluated each article separately. Disagreements were resolved by agreement and discussion with a third party (MB).
Study Selection
Original studies were included if they met the following inclusion criteria: (i) being a randomized controlled trial with either parallel or cross-over design, (ii) investigating the impact of L-carnitine on plasma/serum concentrations of Lp(a), and, (iii) presentation of sufficient information on Lp(a) concentrations at baseline and at the end of follow-up in each group or providing the net change values.
Exclusion criteria were: (i) non-randomized trials, (ii) lack of an appropriate control group in the study design, (iii) observational studies with case-control, cross-sectional or cohort design, (iv) lack of sufficient information on baseline or follow-up Lp(a) concentrations, (v) inability to obtain adequate details of study methodology or results from the article or the investigators, and, (vi) the study was ongoing.
Data extraction
Eligible studies were reviewed and the following data were abstracted: (1) first author’s name, (2) year of publication, (3) study location; (4) study design; (5) number of participants in the L-carnitine and control (in case of randomized design) groups, (6) age, gender and body mass index (BMI) of study participants, (7) baseline levels of total cholesterol, LDL-C, high-density lipoprotein cholesterol (HDL-C), triglycerides, high-sensitivity C-reactive protein (hsCRP) and glucose, (8) systolic and diastolic blood pressures, and, (9) data regarding baseline and follow-up concentrations of Lp(a).
Quality assessment
A systematic assessment of bias in the included studies was performed using the Cochrane criteria38. The items used for the assessment of each study were as follows: adequacy of sequence generation, allocation concealment, blinding and handling of dropouts (incomplete outcome data), selective outcome reporting and other potential sources of bias. According to the recommendations of the Cochrane Handbook, a judgment of “yes” indicated low risk of bias, while “no” indicated high risk of bias. Labeling an item as “unclear” indicated an unclear or unknown risk of bias.
Quantitative Data Synthesis
Meta-analysis was conducted using Comprehensive Meta-Analysis (CMA) V2 software (Biostat, NJ)39. Net changes in measurements (change scores) were calculated as follows: measure at end of follow-up − measure at baseline. For single-arm cross-over trials, net change in plasma concentrations of L-carnitine was calculated by subtracting the value after control intervention from that reported after treatment. Standard deviations (SDs) of the mean difference were calculated using the following formula: SD = square root [(SDpre-treatment)2 + (SDpost-treatment)2 − (2 R × SDpre-treatment × SDpost-treatment)], assuming a correlation coefficient (R) = 0.5. If the outcome measures were reported in median and inter-quartile range, mean and standard SD values were estimated using the method described by Hozo et al.40. Where standard error of the mean (SEM) was only reported, standard deviation (SD) was estimated using the following formula: SD = SEM × square root (n), where n is the number of subjects.
Meta-analysis was performed using either a fixed-effects or random-effect model according to the I2 statistic. I2 values < 50% and ≥ 50% corresponded with the use of fixed-effects and random-effects model, respectively. The generic inverse variance method was used to weight each individual study included in the meta-analysis. Heterogeneity was quantitatively assessed using the I2 index. Effect sizes were expressed as weighted mean difference (WMD) and 95% confidence interval (CI). In order to evaluate the influence of each study on the overall effect size, a sensitivity analysis was conducted using the leave-one-out method (i.e., removing one study each time and repeating the analysis).
Publication bias
Potential publication bias was explored using visual inspection of Begg’s funnel plot asymmetry and Begg’s rank correlation and Egger’s weighted regression tests. Duval & Tweedie’s “trim and fill” method was used to adjust the analysis for the effects of publication bias41.
Results
Search results and trial flow
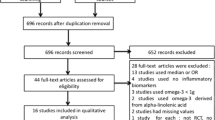
The initial screening for possible interpretation eliminated the articles whose titles and/or abstracts were clearly insignificant. After evaluation, 7 studies with 8 treatment arms (intravenous administration: 2 eligible studies with 3 treatment arms, oral administration: 5 eligible studies with 5 treatment arms) met the inclusion criteria and were chosen for the final meta-analysis. A study flow chart is presented on Fig. 1.

Flow chart of the number of studies identified and included into the meta-analysis.
Characteristics of included studies
In total, 375 participants were randomized, of whom 200 were assigned to the L-carnitine supplementation group and 175 to the control group. The number of participants in these trials ranged from 15 to 46. Studies were published between 2000 and 2014 and were conducted in the Italy, China and Iran. A range of doses from 1 g 3 times per week to 4 g/day of L-carnitine was administered in the included trials. Duration of L-carnitine supplementation ranged from 1 week to 24 weeks. L-carnitine appeared safe and well-tolerated in all RCTs included in this analysis, with no report of any serious adverse events. Demographic and baseline parameters of the included studies are shown in Table 1.
Risk of bias assessment
According to the Cochrane Collaboration, a specific tool for assessing risk of bias in each included study comprises judgment of specific features of the study42. This involves evaluating the risk of bias as ‘low risk’, ‘high risk or ‘unclear risk’. The final category implies either lack of information or doubt over the potential for bias. There are seven analyzed domains comprising: sequence generation (selection bias), allocation sequence concealment (selection bias), blinding of participants and personnel (performance bias), blinding of outcome assessment (detection bias), incomplete outcome data (attrition bias), selective outcome reporting (reporting bias) and other potential sources of bias (Table 2).
Effect of L-carnitine on plasma Lp(a) concentrations
Overall, the impact of L-carnitine on plasma Lp(a) levels were reported in 7 trials comprising 8 treatment arms with 375 participants. Meta-analysis suggested a significant reduction of Lp(a) levels following supplementation with L-carnitine (WMD: −8.82 mg/dL, 95% CI: −10.09, −7.55, p < 0.001). This result was robust in the leave-one-out sensitivity analysis (Fig. 2). When the studies were categorized according to the route of administration, a significant reduction in plasma Lp(a) concentration was observed with oral (WMD: −9.00 mg/dL, 95% CI: −10.29, −7.72, p < 0.001) (Fig. 3) but not intravenous L-carnitine supplementation (WMD: −2.91 mg/dL, 95% CI: −10.22, 4.41, p = 0.436). Both of these latter effects were robust in the sensitivity analysis (Fig. 4).

Forest plot detailing weighted mean difference and 95% confidence intervals for the impact of L-carnitine on plasma Lp(a) concentrations.
Lower plot shows leave-one-out sensitivity analysis.

Forest plot detailing weighted mean difference and 95% confidence intervals for the impact of oral L-carnitine on plasma L(a) concentrations.
Lower plot shows leave-one-out sensitivity analysis.

Forest plot detailing weighted mean difference and 95% confidence intervals for the impact of intravenous L-carnitine on plasma Lp (a) concentrations.
Lower plot shows leave-one-out sensitivity analysis.
Effect of L-carnitine on plasma lipids
Overall, the impact of L-carnitine on plasma total cholesterol, LDL-C, HDL-C and triglycerides levels were reported in 7, 6, 6 and 6 treatment arms, respectively. Meta-analysis suggested a significant reduction in plasma concentrations of total cholesterol (WMD: −9.49 mg/dL, 95% CI: −17.07, −1.92, p = 0.014) and a borderline significant trend in reduction of LDL-C (WMD: −4.67 mg/dL, 95% CI: −9.78, 0.45, p = 0.074) following L-carnitine supplementation (Fig. 5). However, plasma HDL-C (WMD: 0.85 mg/dL, 95% CI: −2.34, 4.05, p = 0.601) and triglycerides (WMD: −2.42 mg/dL, 95% CI: −34.85, 30.01, p = 0.884) were not altered by L-carnitine (Fig. 5).

Forest plot detailing weighted mean difference and 95% confidence intervals for the impact of L-carnitine on plasma lipids.
Meta-regression analysis
Potential associations between the Lp(a)-lowering effects of L-carnitine with dose and duration of supplementation were evaluated using meta-regression analysis. The results suggested that the pooled estimate is independent of L-carnitine dose (slope: −0.30; 95% CI: −4.19, 3.59; p = 0.878) and duration of treatment (slope: 0.18; 95% CI: −0.22, 0.59; p = 0.374) (Fig. 6).

Meta-regression plots of the association between mean changes in plasma Lp(a) concentrations after L-carnitine treatment with dose and duration of treatment.
Publication bias
The funnel plot of the study standard error by effect size (WMD) was asymmetric, suggesting potential publication bias that was also confirmed by the results of Egger’s linear regression test (intercept = 1.08, standard error = 0.41; 95% CI = 0.09, 2.09, t = 2.61, df = 6, two-tailed p = 0.04) (Fig. 7). However, there was no sign of bias according to the Begg’s rank correlation test (Kendall’s Tau with continuity correction = 0.32, Z = 1.11, two-tailed p-value = 0.266). An attempt was made to adjust the effect size by imputing potentially missing studies using “trim and fill” correction. This approach led to the imputation of 3 missing trials on the left side of funnel plot, yielding a corrected effect size of −9.16 mg/dL (95% CI: −10.41, −7.92) (Fig. 7). The “fail-safe N” test showed that 89 studies would be needed to bring the effect size down to a non-significant (p > 0.05) value.

Funnel plot detailing publication bias in the studies reporting the impact of L-carnitine on plasma Lp(a) concentrations.
Open diamond represents observed effect size; closed diamond represents imputed effect size.
Discussion
To our knowledge, the current systematic review and meta-analysis is the first to analyze evidence from RCTs on the efficacy of supplementation with L-carnitine on plasma Lp(a) concentrations. The results, albeit with small numbers of subjects overall, suggest a significant reduction of Lp(a) levels following oral supplementation with L-carnitine.
L-carnitine, a derivate of the amino acid lysine, is found predominantly in red meat, certain fish and trace levels observed in dairy and some plant foods, such as avocado43. The effects of L-carnitine on lipid metabolism have been investigated in many experimental and clinical studies, but the exact mechanisms responsible for L-carnitine lowering effects on Lp(a) levels are not completely understood. However, the reason for these effects could be that L-carnitine, by stimulating fatty acid break-down in mitochondria, might reduce liver production of Lp(a)43. An experimental study has shown that L-carnitine inhibits advanced glycation end products (AGE)-modification of bovine serum albumin more potent than aminoguanidine, a prototype inhibitor of AGE43. In hemodialysis patients, L-carnitine supplementation might play a protective role against vascular injury based on reduction in circulating levels of AGEs44.
Recent studies have showed that oral ingestion of L-carnitine in both animals and humans results in generation of trimethylamine by intestinal microbiota, which is absorbed and then metabolized to trimethylamine-N-oxide (TMAO) by liver flavin monoxygenases (FMOs) (especially FMO3 and FMO1)35,45. Since L-carnitine is contained in red meat, omnivorous humans have higher values of TMAO than vegetarians, explaining why L-carnitine and the microbiota may contribute to the causal link between high consumption of red meat and CV risk35. Recent metabolomics and animal studies showed that TMAO, an intestinal microbiota-dependent metabolite formed from dietary trimethylamine-containing nutrients such as phosphatidylcholine (PC), choline and carnitine, is linked to coronary artery disease pathogenesis, only in the presence of TMA/TMAO generation and intact gut microbiota35,46. The mechanisms by which TMAO raises CV risk and accelerates atherosclerosis are not well understood, but studies have shown that TMAO arising from both choline and L-carnitine supplementation in mice may inhibit reverse cholesterol transport35 and enhance foam cell formation in both arterial and peritoneal cavity47. Direct provision of TMAO into diet has been shown to increase the development of atherosclerosis in animal models, confirming its pro-atherogenic properties47 and multiple clinical studies have confirmed that an elevated TMAO level is associated with increased risks for incident major adverse CV events48.
The levels of TMAO are modulated by hepatic FMO3, which is regulated by a bile acid-activated nuclear receptor named the farnesoid X receptor (FXR)49. The FMOs are known to oxidize sulfur-, phosphorus- and selenium-containing compounds and endogenous amines, including TMA50. Antisense oligonucleotide mediated suppression of FMO3 levels have recently been shown to suppress TMAO levels and to inhibit atherosclerosis in animal models51. Furthermore, FMO3 has been shown to serve as a central regulator of tissue cholesterol balance and to be involved in insulin resistance and dyslipidemic changes in mouse models, suggesting the FMO3 and TMAO pathway as potential mediators of diabetes associated atherosclerosis52,53.
In contrast to the potential dangerous effects in the acceleration of atherosclerosis, mixed results have been reported with L-carnitine supplementation in the immediate setting following an acute myocardial infarction (AMI). While the major RCTs examining L-carnitine supplementation in the immediate post-MI setting showed no clinical benefit, a meta-analysis of L-carnitine intervention studies suggested its potential beneficial effect54. Other studies have suggested beneficial effects of L-carnitine supplementation on inflammatory parameters55, in the secondary prevention of CVD54,56, in diabetes mellitus57, on serum lipid profile in hemodialysis patients58, for adults with end-stage kidney disease on hemodialysis59 or for patients in maintenance hemodialysis60, though detailed analysis of whether the carnitine was provided in oral vs intravenous supplementation, bypassing the gut microbiota, has not been extensively evaluated. In our meta-analysis, L-carnitine significantly reduced total cholesterol and showed borderline significant reduction of LDL-C, while plasma HDL-C and triglycerides were not affected. According to the available data L-carnitine supplementation might also significantly reduce triglycerides levels, but especially when low HDL-C levels are present61. However, the real effects of L-carnitine supplementation, taking into account its above-mentioned positive and possibly negative properties (via TMAO), on the process of atherosclerosis and atheroma plaque merit further investigations.
This meta-analysis has several important limitations and the obtained results should be viewed as suggestive only. Most notable, there were only few eligible RCTs included in this meta-analysis and most of them had a relatively modest number of participants. Moreover, none of the RCTs included have hard clinical outcome endpoints and the duration of the interventions was relatively modest. Furthermore, the included studies were heterogeneous concerning the characteristics of patients and study design (2 of studies on L-carnitine supplementation have been performed in the setting of hemodialysis when carnitine metabolism is altered in uremia and the depletion of carnitine is expected62, 4 in diabetic/metabolic syndrome patients with dyslipidemia and 1 in hyperlipidemic patients). There was also the diversity of the co-administered medications (statins or coenzyme Q10). Moreover the duration of the L-carnitine supplementation differs from one study to another, therefore further studies are required to test the time dependence of the observed effect as well as the impact of L-carnitine bioavailability on the Lp(a)-lowering activity. Many characteristics that vary within studies, such as the type of study, the year of publication, the inclusion and exclusion criteria, the sample size, the control group or quality of the studies, could have been the factors of between-study heterogeneity. However, we used “leave-one-out” sensitivity analysis under a conservative random-effects model to assess the possibility of some bias such as unclear double-randomization or large unbalanced dropout. Our results suggest that the significance of estimated pooled effect size is not biased by any single study.
The meta-analysis suggests that L-carnitine supplementation might be associated with significant Lp(a) lowering. However, given the TMAO elevating effect of oral L-carnitine supplementation, whether or not L-carnitine is a reasonable therapy to reduce Lp(a) levels requires further long-term investigations with hard clinical endpoints such as AMI, stroke and mortality risks. Prospective trials are required to fully elucidate the clinical value of L-carnitine supplementation.
Additional Information
How to cite this article: Serban, M.-C. et al. Impact of L-carnitine on plasma lipoprotein(a) concentrations: A systematic review and meta-analysis of randomized controlled trials.. Sci. Rep. 6, 19188; doi: 10.1038/srep19188 (2016).
References
Marcovina, S. M., Koschinsky, M. L., Albers, J. J. & Skarlatos, S. Report of the National Heart, Lung and Blood Institute Workshop on Lipoprotein (a) and Cardiovascular Disease: recent advances and future directions. Clinical chemistry 49, 1785–1796 (2003).
Ramasamy, I. Recent advances in physiological lipoprotein metabolism. Clinical Chemistry and Laboratory Medicine (CCLM) 52, 1695–1727 (2014).
Clarke, R. et al. Genetic variants associated with Lp (a) lipoprotein level and coronary disease. New England Journal of Medicine 361, 2518–2528 (2009).
Berglund, L. & Ramakrishnan, R. Lipoprotein (a) an elusive cardiovascular risk factor. Arteriosclerosis, thrombosis and vascular biology 24, 2219–2226 (2004).
Lippi, G. & Guidi, G. Biochemical risk factors and patient’s outcome: the case of lipoprotein (a). Clinica chimica acta 280, 59–71 (1999).
Lippi, G., Braga, V., Adami, S. & Guidi, G. Modification of serum apolipoprotein AI, apolipoprotein B and lipoprotein (a) levels after bisphosphonates-induced acute phase response. Clinica chimica acta 271, 79–87 (1998).
Mellwig, K. et al. [Lipoprotein (a): influence on cardiovascular manifestation.]. Clinical research in cardiology supplements 10, 33–8 (2015).
Bennet, A. et al. Lipoprotein (a) levels and risk of future coronary heart disease: large-scale prospective data. Archives of internal medicine 168, 598–608 (2008).
Gurdasani, D. et al. Lipoprotein (a) and Risk of Coronary, Cerebrovascular and Peripheral Artery Disease The EPIC-Norfolk Prospective Population Study. Arteriosclerosis, thrombosis and vascular biology 32, 3058–3065 (2012).
Collaboration, E. R. F. Lipoprotein (a) concentration and the risk of coronary heart disease, stroke and nonvascular mortality. JAMA: the journal of the American Medical Association 302, 412 (2009).
Vongpromek, R. et al. Lipoprotein (a) levels are associated with aortic valve calcification in asymptomatic patients with familial hypercholesterolaemia. Journal of internal medicine 278, 166–73 (2015).
Lippi, G., Franchini, M. & Targher, G. Screening and therapeutic management of lipoprotein (a) excess: review of the epidemiological evidence, guidelines and recommendations. Clinica Chimica Acta 412, 797–801 (2011).
Nicholls, S. J. et al. Lipoprotein (a) levels and long-term cardiovascular risk in the contemporary era of statin therapy. Journal of lipid research 51, 3055–3061 (2010).
Allen, S. et al. Expression of adhesion molecules by Lp (a): a potential novel mechanism for its atherogenicity. The FASEB journal 12, 1765–1776 (1998).
Kang, C. et al. Lp (a) Particles Mold Fibrin-Binding Properties of Apo (a) in Size-Dependent Manner A Study With Different-Length Recombinant Apo (a), Native Lp (a) and Monoclonal Antibody. Arteriosclerosis, thrombosis and vascular biology 22, 1232–1238 (2002).
Tsimikas, S., Tsironis, L. D. & Tselepis, A. D. New insights into the role of lipoprotein (a)-associated lipoprotein-associated phospholipase A2 in atherosclerosis and cardiovascular disease. Arteriosclerosis, thrombosis and vascular biology 27, 2094–2099 (2007).
Nielsen, L. B. Atherogenecity of lipoprotein (a) and oxidized low density lipoprotein: insight from in vivo studies of arterial wall influx, degradation and efflux. Atherosclerosis 143, 229–243 (1999).
Nordestgaard, B. G. et al. Lipoprotein (a) as a cardiovascular risk factor: current status. European heart journal 31, 2844–2853 (2010).
Goldstein, L. B. et al. Guidelines for the primary prevention of stroke a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke; a journal of cerebral circulation 42, 517–584 (2011).
Banach, M. et al. Lipids, blood pressure and kidney update 2014. Pharmacological Research 95, 111-125 (2015).
Lippi, G. & Targher, G. Optimal therapy for reduction of lipoprotein (a). Journal of clinical pharmacy and therapeutics 37, 1–3 (2012).
Kotani, K. et al. Tibolone can decrease lipoprotein(a) concentrations in postmenopausal women: a systematic review and meta-analysis of controlled trials. Eur Heart J 26(Suppl. 1), 286 (2015).
Kotani, K. et al. Tibolone decreases Lipoprotein (a) levels in postmenopausal women: A systematic review and meta-analysis of 12 studies with 1009 patients. Atherosclerosis 242, 87–96 (2015).
Banach, M. et al. PCSK9 inhibition-a novel mechanism to treat lipid disorders? Current pharmaceutical design 19, 3869–3877 (2013).
Dragan, S., Serban, M. C. & Banach, M. Proprotein convertase subtilisin/kexin 9 inhibitors: an emerging lipid-lowering therapy ? J Cardiovasc Pharmacol Ther, 20, 157–168 (2015).
Raal, F. J. et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. The Lancet 375, 998–1006 (2010).
Ladenson, P. W. et al. Use of the thyroid hormone analogue eprotirome in statin-treated dyslipidemia. New England Journal of Medicine 362, 906–916 (2010).
Samaha, F. F., McKenney, J., Bloedon, L. T., Sasiela, W. J. & Rader, D. J. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nature Clinical Practice Cardiovascular Medicine 5, 497–505 (2008).
Merki, E. et al. Antisense oligonucleotide lowers plasma levels of apolipoprotein (a) and lipoprotein (a) in transgenic mice. Journal of the American College of Cardiology 57, 1611–1621 (2011).
Tsimikas S. et al. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet 386, 1472–83 (2015).
Banach, M. et al. Lipid, blood pressure and kidney update 2013. International urology and nephrology 46, 947–961 (2014).
Sahebkar, A., Serban, C., Ursoniu, S. & Banach, M. Effect of garlic on plasma lipoprotein(a) concentrations: A systematic review and meta-analysis of randomized controlled clinical trials. Nutrition. 32, 33–40 (2016).
Broderick, T. L. ATP production and TCA activity are stimulated by propionyl-L-carnitine in the diabetic rat heart. Drugs in R & D 9, 83–91 (2008).
Sanchez-Niño, M. D. & Ortiz, A. Differential effects of oral and intravenous l-carnitine on serum lipids: is the microbiota the answer? Clinical Kidney Journal 7, 437–441 (2014).
Koeth, R. A. et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nature medicine 19, 576–585 (2013).
Koeth, R. A. et al. γ-Butyrobetaine Is a Proatherogenic Intermediate in Gut Microbial Metabolism of L-Carnitine to TMAO. Cell metabolism 20, 799–812 (2014).
Moher, D., Liberati, A., Tetzlaff, J., Altman, D. G. & Group, P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ 339, b2535, 10.1136/bmj.b2535 (2009).
Higgins, J. (Chichester, UK, John Wiley and Sons Ltd. Ref Type: Report, 2010).
Borenstein, M., Hedges, L., Higgins, J. & Rothstein, H. Comprehensive meta-analysis version 2. Englewood, NJ: Biostat 104, (2005).
Hozo, S. P., Djulbegovic, B. & Hozo, I. Estimating the mean and variance from the median, range and the size of a sample. BMC medical research methodology 5, 13 (2005).
Duval, S. & Tweedie, R. Trim and fill: a simple funnel‐plot–based method of testing and adjusting for publication bias in meta‐analysis. Biometrics 56, 455–463 (2000).
Green, S. Cochrane handbook for systematic reviews of interventions version 5.1. 0 [updated March 2011]. The Cochrane Collaboration (2011).
Rajasekar, P. & Anuradha, C. L-Carnitine inhibits protein glycation in vitro and in vivo: evidence for a role in diabetic management. Acta diabetologica 44, 83–90 (2007).
Fukami, K. et al. Oral L-carnitine supplementation increases trimethylamine-N-oxide, but reduces markers of vascular injury in hemodialysis patients. Journal of cardiovascular pharmacology 65, 289–295 (2015).
Rysz, J. et al. Increased levels of soluble TNF-alpha receptors and cellular adhesion molecules in patients undergoing bioincompatible hemodialysis. Am J Nephrol 26, 437–44 (2006).
Wang, Z. et al. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. European heart journal 35, 904–10 (2014).
Wang, Z. et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63 (2011).
Tang, W. H. et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res 116, 448–455 (2015).
Bennett, B. J. et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell metabolism 17, 49–60 (2013).
Tang, W. & Hazen, S. L. The contributory role of gut microbiota in cardiovascular disease. The Journal of clinical investigation 124, 4204–4211 (2014).
Shih, D. M. et al. Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. Journal of lipid research 56, 22–37 (2015).
Warrier, M. et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell reports. 10.1016/j.celrep.2014.12.036.
Miao, J. et al. Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nature communications 6, 6498 (2015).
DiNicolantonio, J. J., Lavie, C. J., Fares, H., Menezes, A. R. & O’Keefe, J. H. L-carnitine in the secondary prevention of cardiovascular disease: systematic review and meta-analysis. Mayo Clinic Proceedings 88, 544–551 (2013).
Sahebkar, A. Effect of L-Carnitine Supplementation on Circulating C-Reactive Protein Levels: A Systematic Review and Meta-Analysis. Journal of Medical Biochemistry 34, Pages 151–159, ISSN (Online) (2015).
Shang, R., Sun, Z. & Li, H. Effective dosing of L-carnitine in the secondary prevention of cardiovascular disease: a systematic review and meta-analysis. BMC cardiovascular disorders 14, 88 (2014).
Vidal-Casariego, A. et al. Metabolic effects of L-carnitine on type 2 diabetes mellitus: systematic review and meta-analysis. Experimental and clinical endocrinology & diabetes 121, 234–238 (2013).
Huang, H., Song, L., Zhang, H., Zhang, J. & Zhao, W. Influence of L-Carnitine Supplementation on Serum Lipid Profile in Hemodialysis Patients: A Systematic Review and Meta-Analysis. Kidney & blood pressure research 38, 31–41 (2014).
Chen, Y. et al. L-Carnitine supplementation for adults with end-stage kidney disease requiring maintenance hemodialysis: a systematic review and meta-analysis. The American journal of clinical nutrition 99, 408–422 (2014).
Yang, S.-k. et al. Effect of L-carnitine therapy on patients in maintenance hemodialysis: a systematic review and meta-analysis. Journal of nephrology 27, 317–329 (2014).
Vacha, G. M., Giorcelli, G., Siliprandi, N. & Corsi, M. Favorable effects of L-carnitine treatment on hypertriglyceridemia in hemodialysis patients: decisive role of low levels of high-density lipoprotein-cholesterol. The American journal of clinical nutrition 38, 532–540 (1983).
Golper, T. A. et al. Multicenter trial of L-carnitine in maintenance hemodialysis patients. I. Carnitine concentrations and lipid effects. Kidney Int 38, 904–911 (1990).
Derosa, G. et al. The effect of L-carnitine on plasma lipoprotein(a) levels in hypercholesterolemic patients with type 2 diabetes mellitus. Clinical therapeutics 25, 1429–1439 (2003).
Galvano, F. et al. Effects of simvastatin and carnitine versus simvastatin on lipoprotein(a) and apoprotein(a) in type 2 diabetes mellitus. Expert opinion on pharmacotherapy 10, 1875–1882 (2009).
Shakeri, A., Tabibi, H. & Hedayati, M. Effects of L-carnitine supplement on serum inflammatory cytokines, C-reactive protein, lipoprotein (a) and oxidative stress in hemodialysis patients with Lp (a) hyperlipoproteinemia. Hemodialysis international 14, 498–504, (2010).
Shojaei, M., Djalali, M., Khatami, M., Siassi, F. & Eshraghian, M. Effects of carnitine and coenzyme Q10 on lipid profile and serum levels of lipoprotein(a) in maintenance hemodialysis patients on statin therapy. Iranian journal of kidney diseases 5, 114–118 (2011).
Sirtori, C. et al. L-carnitine reduces plasma lipoprotein (a) levels in patients with hyper Lp (a). Nutrition, metabolism and cardiovascular diseases 10, 247–251 (2000).
Solfrizzi, V. et al. Efficacy and tolerability of combined treatment with L-carnitine and simvastatin in lowering lipoprotein(a) serum levels in patients with type 2 diabetes mellitus. Atherosclerosis 188, 455–461 (2006).
Zhang, J. J. et al. L-carnitine ameliorated fasting-induced fatigue, hunger and metabolic abnormalities in patients with metabolic syndrome: a randomized controlled study. Nutrition journal 13, 110 (2014).
Acknowledgements
Funding: This meta-analysis was written independently; no company or institution supported it financially. SLH reports support by grants from the National Institutes of Health and Office of Dietary Supplements (P20HL113452 and R01HL103866). No professional writer was involved in the preparation of this meta-analysis.
Author information
Authors and Affiliations
Contributions
C.S.—designed the study, made the literature search, drafted the manuscript; A.S.—designed the study, made the statistical analysis, corrected the draft of the paper; S.U.—made the statistical analysis, drafted the manuscript; D.P.M., P.P.T., S.R.J., P.M., M.J.B., F.A., S.S.M., C.B., G.Y.H.L., K.K.R., J.R. and S.L.H.—discussed the idea of the meta-analysis, corrected the draft of the paper; M.B.—designed the study, discussed the idea of the meta-analysis, made the literature search, drafted the manuscript, submitted the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Serban, MC., Sahebkar, A., Mikhailidis, D. et al. Impact of L-carnitine on plasma lipoprotein(a) concentrations: A systematic review and meta-analysis of randomized controlled trials. Sci Rep 6, 19188 (2016). https://doi.org/10.1038/srep19188
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep19188
This article is cited by
-
Lipoprotein(a)—When to Screen and How to Treat
Current Cardiovascular Risk Reports (2022)
-
The Role of Nutraceuticals in the Optimization of Lipid-Lowering Therapy in High-Risk Patients with Dyslipidaemia
Current Atherosclerosis Reports (2020)
-
Impact of ezetimibe on plasma lipoprotein(a) concentrations as monotherapy or in combination with statins: a systematic review and meta-analysis of randomized controlled trials
Scientific Reports (2018)
-
Lipoprotein(a)—It Is Risky, but What Do We Do About It?
Current Cardiovascular Risk Reports (2018)
-
Comparison of the effects of fibrates versus statins on plasma lipoprotein(a) concentrations: a systematic review and meta-analysis of head-to-head randomized controlled trials
BMC Medicine (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
