
Abstract
The hypoxia-inducible transcription factor (HIF) co-ordinates the response of tumours to low oxygen by stimulating genes involved in metabolism and angiogenesis. HIF pathway activation is associated with decreased progression-free survival and increased mortality; compounds that target this pathway are potential agents for the treatment of a range of solid tumour malignancies. Renal cancers are likely to be particularly sensitive to inhibition of the HIF pathway since ~80% show constitutive activation of HIF. We have previously described the di-substituted naphthalene derivative, CL67, which binds to a G-quadruplex higher-order structure in the HIF promoter sequence in vitro. We show here that CL67 blocks HIF expression leading to inhibition of HIF-transactivation and down-regulation of downstream target genes and proteins in renal carcinoma cell lines and in a mouse xenograft model of renal cancer. This inhibition is independent of pathways that control HIF abundance through oxygen-dependant degradation and oxygen dependant HIF sub-unit expression.
Similar content being viewed by others

Introduction
Areas of reduced oxygen availability (hypoxia) are found in all solid tumours where rapid cellular proliferation without concomitant vascularisation produces a spatially and temporally heterogeneous blood supply1. To meet their increasing metabolic demands and enable growth and progression, tumours must ultimately develop their own vasculature. This process, known as angiogenesis, is one of the hallmarks of cancer and is co-ordinated largely by the pattern of gene expression brought about by hypoxia-inducible factor (HIF) in response to low oxygen levels2,3. Levels of HIF are elevated in a number of human cancers, an observation that is linked to increased progression and mortality, making inhibitors of the HIF pathway attractive targets for the development of new anti-cancer therapeutics4.
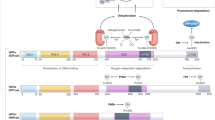
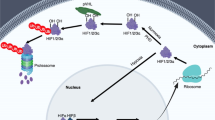
HIF-1 is a heterodimer consisting of a constitutively expressed HIF-1β subunit and an oxygen-dependent HIF-α subunit that together bind specific hypoxia response element (HRE: 5′-(A/G)CGTG-3′) sequences in promoters of their target genes5. There are three α isoforms; HIF-1α plays a general role in hypoxia signaling while HIF-2α and HIF-3α show a more context specific pattern of expression6,7. Control of HIF levels and activity is achieved through expression and degradation of the α subunits by oxygen dependent and independent mechanisms (Fig. 1a). The principal oxygen-dependant control of HIF levels is mediated by the von Hippel Lindau (VHL) protein. In normoxia (21% oxygen), conserved prolines 402 and/or 564 are hydroxylated by prolyl hydroxylase domain (PHD) enzymes which require O2 as a substrate. Hydroxylated HIF-α subunits are recognised by VHL which in turn recruits an ubiquitin ligase complex via Elongin C, directing HIF-α to proteosomal degradation8. Under continuous hypoxia, HIF-α subunits remain un-hydroxylated, are not recognised by VHL and can therefore form stable heterodimers with HIF-1β subunits. Dimerised HIF proteins are then able to bind their cognate HREs and co-ordinate the transcription of pro-angiogenic growth factors such as vascular endothelial growth factor (VEGF), as well as numerous genes that contribute to cancer progression including Angiopoietin-like 4 (ANGPTL4)9. In the absence of prolyl hydroxylation (i.e. in an oxygen independent manner) the RACK1 protein can also interact with HIF-α to recruit Elongin C leading to HIF ubiquitylation and proteolysis. This interaction is inhibited by HSP90 which competes with RACK1 for the same binding site on HIF-1α10. Cellular levels of active HIF are also stimulated by the phosphatidylinositide-3-kinase(PI3K)/AKT/mammalian target of rapamycin (mTOR) and mitogen-activated protein kinase (MAPK) pathways that converge on p70 S6 kinase and eIF-4E binding proteins, leading to increased translation of HIF-α mRNAs11,12,13.

Regulation of the HIF pathway by oxygen dependant and independent mechanisms (a) Under normoxia, prolyl hydroxylase domain proteins use oxygen and α-ketoglutarate as substrates to hydroxylate conserved proline residues on HIF-α subunits. This allows binding of the VHL protein which recruits an ubiquitin ligase complex via Elongin C, leading to HIF-α ubiquitination and proteosomal degradation8. Oxygen-independent control of HIF is mediated by RACK1 which also recruits Elongin C and directs the HIF-α subunits to the proteasome. PI3K/AKT/mTOR and MAPK pathways can increase translation of HIF-α mRNA in an oxygen-independent manner11. In hypoxia, stabilised HIF-α subunits dimerise with HIF-β subunits enabling interaction with HREs in a wide range of gene promoters in the nucleus. The resulting pattern of transcription drives cellular responses to hypoxia such as angiogenesis and metabolic reprogramming and can up-regulate the expression of multiple genes that contribute to cancer progression through survival, metastasis and invasion13. (b) Structure of CL67.
Genetic and immunohistochemical analyses of different populations have reported between 50% and 91% of familial and sporadic renal cell carcinomas are associated with genetic or epigenetic abnormalities within VHL that cause loss of function and are associated with increased expression of HIF-1α and HIF-2α14,15,16,17,18,19. Constitutive expression of the HIF pathway is commonly regarded as the foremost oncogenic cause of renal cancers and inhibitors of the HIF pathway are likely to be particularly effective in treating renal cell carcinoma. Current molecular strategies to inhibit the HIF pathway include direct inhibition of HIF-1α using anti-sense RNA20, blockage of HIF-1α/HIF-1β dimerization21 and re-purposing of existing anticancer agents that inhibit components of the PI3K/AKT/mTOR and MAPK22,23,24,25. While most work has focused on targeting the HIF-1α subunit, HIF-2α is also commonly up-regulated in renal cancers26, where it is specifically responsible for the up-regulation of pro-tumorigenic genes27,28.
Our laboratory's approach to the discovery of HIF inhibitors involves targeting promoter sequences containing several short runs of guanine nucleotides that can form complex higher order structures, termed quadruplexes29. The highly distinctive nature of quadruplex topologies suggests that they can act as novel therapeutic targets, for example in the selective inhibition of transcription of a given oncogene, using designed small molecules to stabilise a particular quadruplex30. The promoters of both HIF-1α and HIF-2α contain guanine-rich sequences capable of forming a G-quadruplex structure one of which has been shown to modulate the activity of the HIF-1α promoter (5′ – GCGCGG4AGG4AGAG5GCG3AGCGCG – 3′), though the role of potential G-quadruplex sequences within the HIF-2α promoter is unknown31,32.
We have identified the G-quadruplex stabilising agent CL67 (Fig. 1b) as a potent and selective inhibitor of renal carcinoma cells in vitro compared to other cancer cell lines including breast, prostate, pancreatic and lung cancer cells33. Here we show that CL67 inhibits both HIF-1α and HIF-2α proteins in a dose- and time-dependent manner both in vitro and in vivo leading to inhibition of HIF-1 transactivation and reduced transcription of downstream target genes.
Results
CL67 inhibits the HIF pathway in renal cancer cell lines
We have recently shown that CL67, a DNA quadruplex-stabilising disubstituted naphthalene derivative, selectively inhibits the growth of renal and osteosarcoma cell lines. In U20S osteosarcoma cells this activity is accompanied by a reduction of HIF-1α and HIF-2α protein levels33. To assess whether CL67 also inhibits the HIF pathway in renal cancer cells, HIF and VEGF protein levels and hypoxia response element (HRE) transactivation were measured in renal clear cell carcinoma lines RCC4 and 786-0 following compound exposure (Fig. 2).

CL67 inhibits the HIF pathway.
(a) Effect of CL67 on HIF protein expression levels in renal cancer cell lines. Left-hand panel: RCC4 cells were incubated in normoxia (21% oxygen) and 786-0 cells in hypoxia (1% oxygen), for 4 h, with the indicated doses of CL67. Right-hand panel: RCC4 cells were treated with five times the IC50 of CL67 for the indicated times. Cells were then lysed and Western blotting was performed probing for HIF-1α, HIF-2α, HIF-1β. Lamins A and C were probed as loading controls. Results are representative of at least 2 independent experiments. (b) CL67 inhibits transcription from the hypoxia response element (HRE). RCC4 cells were co-transfected with either an empty vector control (pGL3; grey bars) or a vector expressing luciferase under the control of multiple copies of the (HRE; red bars). To normalize for transfection efficiency both sets of cells were transformed with a vector expressing renilla luciferase. Cells were then exposed to either normoxia or hypoxia and treated for 4 h with the indicated dosages of CL67. Luciferase activity was measured as described in Methods and final results were expressed as a percentage of the activity of untreated, normoxic cells. (c) CL67 treatment reduces VEGF protein levels in a time- and dose- dependant manner. RCC4 cells were exposed to CL67 at 5× the IC50 for 0, 2 and 4 h (left-hand panel), or for 4 h at 0, 1×, 5× and 10× the IC50 (right-hand panel) Samples (1 ml) of the culture medium were then collected and VEGF levels were measured using ELISA. VEGF levels in all treated samples were significantly reduced compared to untreated controls (p ≤ 0.01). Data in (b) and (c) show the mean ±S.D. of three experiments carried out with at least 2 replicates. Note that cropped blots are shown here.
A dose- and time-dependent decrease in HIF-1α abundance was observed in RCC4 cells in both normoxic and hypoxic conditions (Fig. 2a). Levels of HIF-1α protein in hypoxic RCC4 cells were reduced to 50% of untreated control levels within 2 h of treatment with 5× IC50 of CL67 and reached maximal inhibition (12% of untreated control) after 4 h treatment. There was a marked recovery of HIF-1α levels by 8 h post exposure indicating that this HIF inhibition is reversible. In hypoxic 786-0 cells increasing concentrations of CL67 resulted in a steady decrease (to 61%, 51% and 37% of untreated controls after 4 h treatment with 1×, 5× and 10× IC50CL67, respectively) of the HIF-2α protein (Fig. 2a). It should be noted that 786-0 cells constitutively express HIF-2α but do not express HIF-1α34. No change in the levels of HIF-1β was detected in either cell line.
To confirm the functional relevance of the reduced HIF protein expression, we tested whether CL67 treatment reduced HIF-mediated trans-activation of the HRE using a luciferase reporter construct. In untreated cells HRE trans-activation was increased to 236% of normoxic levels by exposure to hypoxia for 4 h (Fig. 2b). CL67 treatment produced a dose-dependent decrease in HIF activity in RCC4 cells under both normoxic and hypoxic conditions. HRE activity was reduced to approximately baseline (normoxic) levels by 4 h treatment with 1× IC50 CL67 under continued hypoxia and this was significantly reduced to 23.4% and 14.8% of hypoxic untreated control levels by treatment with 5× and 10× IC50 CL67, respectively (p < 0.01). HIF trans-activation was not significantly reduced by treatment with 1× IC50 CL67 under normoxia but was significantly decreased by treatment with 5× and 10× IC50 CL67 (59% and 25% respectively; p < 0.01). CL67 did not have a significant effect on trans-activation of the pGL3 control vector, which lacks the HRE. Further confirmation of the functional relevance of decreased HIF protein levels was determined by measuring the effect of CL67 on the abundance of the HIF down-stream target, VEGF, using ELISA (Fig. 2c). CL67-treated RCC4 cells showed a significant dose- and time-dependent reduction in VEGF abundance (71.6% that of untreated controls within 4 h of treatment with 5× IC50; p < 0.01).
Together these results demonstrated the functional relevance of CL67-mediated inhibition of HIF production, showing that reduced HIF protein levels resulted in reduced activity from HIF-inducible promoters and reduced expression of HIF target proteins.
CL67 inhibits the HIF pathway independently of VHL and upstream oncogenic signalling pathways
Cellular levels of HIF-1α are controlled principally by oxygen-dependant degradation mediated by VHL and oxygen-independent protein synthesis controlled through PI3K-AKT-mTOR and MAP kinase activation13,34,38. To determine if inhibition of HIF by CL67 was achieved through disruption of these pathways, colony formation and the abundances and activities of key members of each pathway were assessed in VHL positive and negative RCC4 cells treated with CL67 (Fig. 3).

CL67 inhibits colony formation and HIF-1α in a manner independent of VHL and the PI3K/AKT/mTOR and MAPK pathways.
(a) RCC4 cells (lacking VHL; RCC4) and RCC4 cells stably transfected with VHL (RCC4/VHL) were incubated for 4 h with the doses indicated of CL67 in normoxia (20% oxygen) or hypoxia (1% oxygen). Colony formation assays were then performed. Data represents the mean ±S.D. of 3 experiments with at least 2 replicates. *, Significant difference compared to untreated control (p < 0.01); **, Significant difference compared to both normoxic sample and untreated sample (p < 0.01). (b and c) RCC4 cells (pVHL −/−) and RCC4 cells stably transfected with pVHL were incubated with 5× IC50 doses of CL67 for the indicated times in normoxia or hypoxia. RCC4/VHL cells were exposed to hypoxia for 16 h to increase levels of HIF-1α before treating with CL67. Cells were lysed and Western blotting was performed probing for HIF-1α (b), HSP-90, Raf-1 and AKT (c). Actin or lamin A were used as loading controls. Data are representative of at least 2 independent experiments. Note that cropped blots are shown here.
RCC4 cells were stably transfected with wild type VHL and exposed to 5× IC50 CL67 under either normoxic or hypoxic conditions. In normoxic cells, restoration of active VHL resulted in a depletion of HIF-1α. This effect was reversed in hypoxic conditions confirming the functionality of the transfected VHL (Fig. 3b). CL67 significantly reduced colony formation in both RCC4 and RCC4/VHL cells after 4 h treatment with 5 × IC50 and 10× IC50 CL67 in both normoxia and hypoxia (Fig. 3a). Furthermore, a time-dependent reduction of HIF-1α protein levels was observed in both normoxic VHL −/− cells (Fig. 3b) and in hypoxic cells carrying wild-type VHL (Fig. 3c). These results demonstrate that CL67 does not depend on the VHL pathway to inhibit colony formation and that CL67 inhibits HIF-1α protein levels by a VHL-independent mechanism.
No significant difference in colony formation was seen in VHL negative RCC4 cells in hypoxia compared to normoxia. Interestingly however, RCC4/VHL cells were significantly less sensitive to CL67 than VHL negative RCC4 cells and were significantly more sensitive to CL67 in hypoxia compared to normoxia (Fig. 3a). The restoration of active VHL also increased IC50 values for CL67 in RCC4, RCC10 and 786-0 renal cell lines (Supplementary Figure S1). Taken together these observations indicate that increasing HIF levels may in fact potentiate the anti-proliferative activity of CL67.
Maximum reduction in HIF-1α levels were observed after 4 h treatment in both VHL positive and negative cell lines. However, protein levels in both lines recovered to between 85% and 91% of the untreated control by 8 h, indicating that the compound is either inactivated (e.g. through metabolism or degradation), or that the CL67-mediated inhibition of HIF production is circumvented by activation of alternate pathways. We therefore measured the abundance of three key proteins, HSP90, AKT and Raf-1, which act in upstream HIF activation pathways (Fig. 3c). None were affected by CL67 treatment in RCC4 cells. In addition, a panel of 65 kinases, including PI3K, MAPK and mTOR, tested negative (IC50 > 10 μM) for inhibition by CL67 (experiments performed by Novartis Institutes for Biomedical Research). A description of the assessed kinases is given in Supporting Table S1. These results indicate that CL67 inhibits HIF independently of both oxygen-dependant and oxygen-independent signalling pathways that are known to control its cellular levels and activity, as well as independently of multiple oncogenic signalling pathways.
CL67 inhibits the expression of HIF-1α and HIF-1 regulated genes
We have demonstrated in vitro that CL67 binds to and stabilises a G-quadruplex sequence located within the promoter sequence of HIF-1α33. We therefore hypothesised that CL67 inhibits the HIF pathway by preventing transcription of the gene as a consequence of stabilising the promoter G-quadruplex sequence. To test this we analysed the mRNA abundance of HIF-1α by qRTPCR using two sets of HIF-1α primers. Using the first primer, set treatment of RCC4 cells with 1×, 5× and 10× IC50 of CL67 reduced HIF-1α mRNA abundance to 29.2%, 29.2% and 27.3% that of the untreated controls respectively (p < 0.05, Student's t-test). In a separate time course, exposure of RCC4 cells to 5× IC50 of CL67, HIF-1α mRNA levels were reduced to 3.9%, 4.0% and 4.2% of the corresponding untreated control after 2, 4 and 6 hours (p < 0.05). In contrast, measurement of the HIF-2α transcript in 786-0 cells by qRTPCR revealed a modest reduction in expression to between 92.5% and 91.4% that of the untreated control following 4 hours exposure to 5× IC50 CL67.
The HIF-1 pathway is a major regulator of angiogenesis in hypoxic tumours39. As a second assessment of HIF-1 pathway blockage and of HIF-1α transcriptional inhibition we measured the expression of 83 genes involved in angiogenesis, in normoxic RCC4 cells. This gene set included HIF-1α (measured with a different primer set than the first experiment), 15 genes that have been robustly shown to be under the control of HIF-1, a further 9 genes that have been linked to the HIF pathway but not demonstrated to be under HIF-1 control and 59 genes that have not been linked to HIF-mediated regulation. The full gene list and dataset for this analysis is given in Supporting Table S2.
There was a titratable decrease in HIF-1α mRNA abundance in normoxic RCC4 cells exposed to 5× IC50 of CL67 for 2 h, 4 h or 6 h compared with untreated cells at 0 h (Fig. 4a). However, in contrast to the first qPCR assay, none of these decreases were strong or statistically significant. Transcripts for all of the 15 genes that are induced by HIF-1 were decreased in abundance over the course of the experiment. The most heavily down-regulated genes were those encoding ANGPTL4, FN1, PLAU and VEGFA, all of which showed statistically significant down-regulation at 6 h exposure (Fig. 4b). Within the angiogenesis gene set there was also a significant reduction in the expression of the 15 HIF-regulated genes compared with the 59 HIF-1 independent genes (Fig. 4c, Supplementary Table S2), indicating that the majority of the observed transcriptional changes were a result of HIF pathway inhibition. Interestingly, there was also a significant down-regulation of the genes encoding tumour necrosis factor-α (TNFα), the chemokines CXCL1 and CXCL6 and the interleukins IL6 and IL8, none of which have previously been shown to be under control of HIF-1 (Fig. 4d). This suggested that these genes could be as yet unidentified targets of the HIF pathway. Alternatively, they could be additional drug targets that are directly inhibited by CL67.

CL67 inhibited the expression of HIF-1α and HIF-1 regulated genes.
(a), (b) and (d)Total RNA was purified from RCC4 cells exposed to 5× the IC50 of CL67 for 2, 4 or 6 hours, reverse transcribed to cDNA and then used to monitor the expression changes of 83 genes involved in angiogenesis by qPCR. Expression data for each gene was normalised to the housekeeping gene RPLP0 and then to untreated samples at t0 (n = 3; error bars, 90% confidence interval; *, p < 0.05 and **, p < 0.01 compared with untreated control using Student's t-test). (c) HIF-1 regulated angiogenesis pathway genes were significantly down-regulated by CL67 compared to HIF-1 independent genes. Shown are box and whisker plots of log2 fold expression changes for 59 genes that are not controlled by the HIF-1 pathway (Non-HIF-1) and 15 genes that are established targets of HIF-1 (HIF-1). Significance values are for 2-tailed, homoscedastic t-tests comparing non-HIF-1 and HIF-1 regulated genes at each time point.
These results further demonstrate that CL67 is a robust inhibitor of the HIF pathway in renal cancer cell lines, inhibiting expression of HIF-regulated genes. This data also indicates that CL67 inhibits the HIF pathway at least in part by inhibiting transcription of HIF-1α, though given the variability of the reduction in the HIF-1α transcript between experiments, other possible mechanisms of inhibition cannot be fully ruled out.
CL67 inhibits the HIF pathway in vivo
We confirmed the in vivo relevance of the HIF inhibitory activity of CL67 by measurement of the expression of both HIF-2α and GLUT-1 proteins in 786-0 cells grown as xenografts on nude mice (Fig. 5). Both proteins were significantly reduced within 6 h of treatment with 15 mg/kg CL67. HIF-2α protein was further reduced to 13.8% of control levels (p < 0.01) after 8 h and GLUT-1 protein to 9.3% of control levels after 16 h (p < 0.01), further confirming the physiological relevance of the HIF inhibitory activity of CL67. As seen in with cultured cells, levels of HIF-2α protein eventually recovered, here after 16 h treatment. RCC4 and RCC4VHL cells could not be grown as xenografts and 786-0 cells do not express HIF-1α so expression of this protein was not assessed in this experiment.

CL67 inhibits the HIF pathway in vivo.
(a) 786-0 cells were grown as xenografts in the flanks of nude mice. Once the xenografts had reached an appropriate size (median 150 mm3) mice were treated with 15 mg/kg CL67. Tumours were harvested from four mice at each time point and were immediately homogenised and lysed on ice. Western blotting was then performed with antibodies against HIF-2α, GLUT-1, lamin A and lamin C. Representative blots for two of the four mice at each time point are shown. A common sample from the untreated control group was run on each gel as a reference to enable comparison between gels. (b) Blots were quantified using ImageQuant Software, normalised for loading and expressed as a percentage of the intensity of the common reference sample (Data are means ± s.e.; n = 4, *, p < 0.05 compared with the control group). Note that cropped blots are shown here.
Discussion
Up to 91% of renal cancers over-express HIF-1α due to inactivation of VHL14,15,16,17,18,19 and are therefore likely to be over-reliant on this pathway for their genesis, growth and survival. We have previously shown that CL67 is a novel small molecule inhibitor that preferentially inhibits renal cancer cell line growth compared to other cancer cell lines including human breast, lung, prostate and pancreatic cell lines33. These studies also demonstrated that CL67 binds to and stabilises G-quadruplex sequences in the HIF-1α and HIF-2α promoter sequences in vitro, using surface plasmon resonance33. The current study provides the first evidence that a small molecule G-quadruplex stabilising agent can produce inhibition of both HIF-1α and HIF-2α proteins with subsequent down-regulation of the HIF pathway.
Renal cell carcinoma cells exposed to CL67 displayed decreased levels of HIF-1α and HIF-2α proteins and decreased transactivation of the phosphoglycerate kinase HRE in both normoxia and hypoxia and in both the presence and absence of VHL (Fig. 2 and Fig. 3), demonstrating a robust inhibitory effect that is likely to be independent of both tumour oxygen status and VHL. Down-stream target proteins of the HIF pathway, VEGF and GLUT-1 were also decreased following drug treatment. Though VEGF protein levels were not reduced to as great an extent as HIF-1α and HIF-2α proteins or HRE trans-activation, the values obtained were comparable with previous studies that have shown similar effects on VEGF levels following inhibition of HIF-1 and which were attributed to VEGF production through compensatory HIF-independent mechanisms40. A qPCR array analysis of angiogenesis pathway genes revealed that CL67 also significantly down-regulated HIF-regulated genes compared with genes that are not under HIF control. Amongst the genes down-regulated by CL67 were ANGPTL4, EDN1 and VEGFA, all of which contribute to cancer progression13. Interestingly, pro-inflammatory cytokines IL6 and TNFα were also significantly down-regulated in a time-dependent manner by CL67 treatment. Activation of the IL6 receptor is known to induce HIF-1α expression41, indicating that the observed IL6 transcriptional inhibition could be linked with CL67-mediated blockage of the HIF pathway.
Several signalling pathways are known to regulate HIF activity and modulation of these can account for the down-regulation of HIF by small molecules. For example, inhibitors of HSP90 induce HIF-1α ubiquitination via RACK1 leading to protein degradation even in the absence of VHL (Fig. 1a10,42). HSP90 levels in RCC4 cells were unaffected by the CL67 dosages and exposure times that resulted in depletion of HIF-1 proteins. Similarly, CL67 did not affect the expression of Raf-1 and AKT, key members of the MAPK and PI3K/AKT/mTOR pathways respectively that control HIF-1α protein expression44, nor did they inhibit the activity of a kinase panel. Though we cannot fully rule out other non-specific effects, the current data support the conclusion that CL67 is a specific and direct inhibitor of the HIF pathway.
In interesting observation was the decreased capacity of CL67 to inhibit colony formation and in vitro growth of renal carcinoma cells in which active VHL had been restored (Fig. 3a and supplementary data). Due to inactive VHL, RCC4 cells express high levels of HIF-1α in both normoxia and hypoxia. The differential effects of CL67 on VHL-positive and VHL-negative cell lines therefore indicate that increased HIF expression potentiates the anti-proliferative activity of CL67 and therefore has positive implications for the ability of this compound to selectively inhibit the growth of carcinomas in which the HIF pathway is over-active.
There was a notable discrepancy between the qPCR data obtained using a qPCR array, which showed a titratable but insignificant decrease in HIF-1α mRNA (Fig. 4a) and the values obtained using specific qPCR primers that indicated a strong and significant down-regulation of HIF-1α mRNA (reduced to between 3.9% and 29.2% of untreated control). Furthermore, HIF-2α mRNA in 786-0 cells was not significantly reduced despite strong down regulation of HIF-2α protein in 786-0 tumour xenografts (Fig. 5). We cannot therefore safely conclude that CL67 acts as a transcriptional inhibitor of HIF-1α or HIF-2α. One alternative explanation is that stabilisation of an RNA G-quadruplex within the HIF-1α 5′UTR could be responsible for the drop in protein levels. This mechanism would be consistent with the recovery of HIF levels we observed both in cells and in vivo as the continued production of HIF transcripts could titrate out cellular CL67, allowing subsequent restoration of expression. These questions will drive future work into the molecular mechanism of CL67.
The HIF-1α promoter contains a polypurine/polypyrimidine tract (−65 to −85), which has been previously identified as forming a parallel, intramolecular G-quadruplex31. Site-directed mutagenesis of this region markedly reduces HIF-1α transcriptional activity, whilst two G-quadruplex ligands, telomestatin and TmPyP4, have been shown to bind and stabilise the G-quadruplex formed by the HIF-1α polypurine sequence31. Interestingly, it was also noted that the HIF-2α promoter contains a nearly identical polypurine/polypyrimidine tract which appears capable of forming an intramolecular G-quadruplex. Our current data together with previous work33 suggests, though does not prove, that G-quadruplex in both HIF-1α and HIF-2α are targeted by CL67 and further studies are needed to elucidate the precise mechanisms and targets involved. However, since the polypurine/polypyrimidine tract that is likely to be able to form an intramolecular G-quadruplex differs from the HIF-1α quadruplex sequence by only two bases31, it seems likely (though not proven) that the inhibition of HIF-2α observed in the current study is also a result of CL67 binding to a G-quadruplex sequence in HIF-2α. In addition, the polypurine tract involved in G-quadruplex formation contains binding sites for the activating transcription factors Sp1 and AP231 and we cannot rule out that CL67 may affect their binding thereby altering transcription of HIF-1α and HIF-2α. However, our data demonstrating that CL67 inhibits both HIF-1α and HIF-2α mRNA, in addition to the data showing that CL67 acts independently of the VHL, PI3K/AKT/mTOR and MAPK pathways, provides additional evidence that CL67 inhibits the HIF pathway via a novel mechanism distinct from any previous HIF inhibitor.
It is thought that HIF-1α and HIF-2α may play different roles in the cellular response to hypoxia and nutrient deprivation depending on their cellular environment and that their responses may be somewhat cell specific43. Interestingly, the expression of both HIF-1α and HIF-2α varied within the cell lines used during this study but the effect on the downstream HIF pathway (transactivation and downstream gene expression) after inhibition of HIF-1α and HIF-2α by CL67 was similar. CL67 also inhibits cell growth to a similar extent in a panel of renal cell lines with varying expression of HIF-1α and HIF-2α (IC50 values of 1.4–2.6 μM)33. Long-term growth studies in animal models may help to elucidate the relative effects of inhibition of HIF-1α and HIF-2α more fully.
In summary, we have identified CL67 as a potent inhibitor of the HIF pathway which we propose acts by binding to and stabilising G-quadruplex structures in HIF-1α and HIF-2α leading to decreased transcription, down-regulation of HIF-1α and HIF-2α proteins and inhibition of HIF transactivation with subsequent reduction in expression of down-stream target genes, both in vitro and in vivo. In addition, the compound also down-regulates expression of pro-inflammatory cytokine IL6. CL67 therefore represents an exciting therapeutic agent, with a novel mechanism of action, which may offer therapeutic advantage in the treatment of human renal cancers.
Methods
Cell culture and drug treatments
A498 and 786-0 human renal carcinoma cells were obtained from ATCC. RCC4 and RCC4VHL (RCC4 cells transfected with the functional VHL gene34,35) human renal carcinoma cells were obtained from the European Collection of Cell Cultures. Cells were cultured as recommended by suppliers in either DMEM, MEM or RPMI supplemented with 2 mM glutamine and 10% Foetal Bovine Serum. RCC4 and RCC4VHL cells were also maintained in 0.5 mg/ml G418 (Invitrogen). Growth and drug treatments were performed in humidified air, 21% O2, 5% CO2, 74% N2 (normoxia) or 1% O2, 5% CO2, 94% N2 (hypoxia) using an In Vivo Hypoxia Workstation 400 (Ruskinn, UK). Analytically pure CL67 was dissolved in DMSO. Cells treated with CL67 at increasing concentrations (1×, 5× or 10× IC50) and/or exposure times (0 to 8 h) were washed in ice-cold PBS, harvested by scraping into 10 ml ice-cold PBS and collected by centrifugation (300 g, 3 min, 4°C) prior to re-suspension into the relevant assay buffer. IC50 values for CL67, reported previously33 were 1.4 μM in RCC4 cells and 2.3 μM for 786-0 cells.
Western blotting
Western blotting was performed as described previously36 using mouse anti-human HIF-1α (1:250; Transduction Labs), HIF-2α goat anti-human HIF-1β (1:200; Santa Cruz), goat anti-human lamin A/C (1:500; Santa Cruz), rabbit anti-human AKT (1:1000; Cell Signalling Technology), rabbit anti-human HSP90 (1:1,000; Santa Cruz), goat anti-human actin (1:1,000; Santa Cruz), mouse anti-human Raf-1 (1:1,000; Santa Cruz) and mouse anti-human GLUT-1 (1:200; Santa Cruz). All blots were incubated in primary antibodies overnight at 4°C with shaking. Appropriate secondary antibodies were used at 1:5,000 (1 h, room temperature with shaking). Blots were quantified using ImageQuant software (Molecular Dynamics).
Luciferase reporter assays
A pGL3 firefly luciferase reporter plasmid containing the phosphoglycerate kinase HRE was supplied by Professor P. Maxwell (University College London, UK)34. The empty pGL3 control plasmid and the pRL-CMV Renilla luciferase plasmids were obtained from Promega. Cells were transfected with the pRL-CMV Renilla luciferase plasmid and either the HIF-1 reporter plasmid or the pGL3 control plasmid using LipoTAXI (Stratagene) and 24 h later were exposed to normoxia or hypoxia for the required time, with or without CL67. Firefly and Renilla luciferase activities were measured using the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions.
VEGF measurements
Approximately 107 cells were treated with CL67 as required. 1 mL of cell culture medium was removed and cleared using centrifugation (1,500 r.p.m., 4°C, 3 min). The supernatant was stored at −80°C prior to use. VEGF abundance was quantified using a human VEGF ELISA kit according to the manufacturer's instructions (R&D Systems, Minneapolis, MN).
Clongenicity assays
RCC4 cells (lacking VHL; RCC4) and RCC4 cells stably transfected with VHL (RCC4/VHL) were incubated for 4 h with the doses indicated of CL67 in normoxia (20% oxygen) or hypoxia (1% oxygen) in T-25 flasks containing 5 ml medium. Cells were then washed three times in 10 ml PBS and trypsinised using standard protocols. 100, 500 and 1,000 cells were plated in duplicate into 6-well plates containing 10 ml medium and were incubated in normoxia for 14 days. Cells were then washed three times in 10 ml PBS, fixed using 5 ml 70% ethanol and colonies containing >50 cells were counted after staining (2% methylene blue in 50% ethanol, 2 ml). Results were expressed as a percentage of untreated controls and represent the mean of three experiments carried out in duplicate.
Reverse transcriptase PCR (RT-PCR)
Total RNA was isolated from approximately 107 cells using the RNeasy kit (Qiagen, Crawley, UK) according to the manufacturer's protocol. HIF-1α transcripts were quantified using two-step, quantitative real time PCR (qRTPCR). First-strand cDNA synthesis was performed for each sample with a Protoscript M-MuLV Taq RT-PCR kit (New England Biolabs, MA, USA) using 1 μg total RNA and oligo-dT primers. qPCR was performed using Stratagene Brilliant III SYBR Green master mix in a Stratagene MX-3000P instrument (Agilent, TX, USA) according to the manufacturer's instructions using 2 ng input cDNA and primers at 300 nM. The following primer sequences were used: HIF-1α forward: CCAGTTACGTTCCTTCGATCAGT; HIF-1α reverse: TTTGAGGACTTGCGCTTTCATCA, HIF-2α forward: AAGCCTTGGAGGGTTTCATTG, HIF-1α reverse: TGCTGATGTTTTCTGACAGAAA, β-actin forward: CCCAGAGCAAGAGAGG, β-actin reverse: GTCCAGACGCAGGATG. Duplicate biological samples were measured in duplicate for each gene, then fold-regulation relative to the untreated control was determined using the Relative Expression Software Tool–384, version 237. Samples were normalised to β-actin. Differences in PCR efficiency were normalised using relative standard curves of cDNA pooled from all samples. P-values were determined for 2000 randomisations.
Angiogenesis pathway analysis
The expression of 83 genes involved with angiogenesis was assessed using human angiogenesis PCR arrays (SABiosciences). Total RNA was extracted from CL67-treated cells using the RNeasy kit with on-column DNaseI treatment (Qiagen) according to the manufacturer's protocol. The quality of the harvested RNA was confirmed by nanodrop spectroscopy and by analysis with a Human RT2 RNA QC PCR Array. First strand cDNA synthesis was performed with the RT2 first strand kit and qPCR was carried out using the RT2 qPCR master mix (Qiagen) as described above. Relative gene expression was determined by the ΔΔCT method using the web-based RT2 Profiler™ PCR Array Data Analysis software (SABiosciences). Data were normalised to the geometric mean of the house keeping gene RPLP0.
Xenograft studies
Five to six week year old female CD1 and immunodeficient Swiss nude mice were obtained from Harlan UK Ltd and Charles River UK Ltd, respectively. Animals were maintained in individually ventilated caging systems in groups of five at 19°C to 23°C, with a 12-hour light-dark cycle and fed with a conventional diet. All experiments were approved by the institutional Animal Care and Use Committee of The School of Pharmacy London and experimental work was carried out in accordance with UK Home Office regulations.
A dose-range assay was initially performed in female CD1 mice, with an initial mean weight of 25 g. CL67 was dissolved in sterile dH2O and administered intra-peritoneally to the mice (n = 2). The body weight was recorded daily and the animals were observed for clinical symptoms. The starting dose of 0.08 mg/kg was increased step-wise up to 15 mg/kg. No adverse effects of CL67 were observed.
Pharmacodynamics of CL67 in 786-0 xenografts
786-0 tumours were established in the right flank of five to six weeks old immunodeficient Swiss nude mice, by subcutaneous injection of 1 × 107 cells in a mixture of 50:50 (v/v) medium and Matrigel® (BD Bioscences). Tumour development was monitored by serial calliper measurement. Four weeks after inoculation (median tumour volume = 150 mm3) animals were randomly assigned into 6 groups (n = 4). One group was used as a control (untreated) group and the rest of the mice were given CL67 intraperitoneally at a dose of 15 mg/kg. Treated animals were sacrificed at 2 h, 4 h, 6 h, 8 h and 16 h after injection. The tumour tissue was immediately excised and placed into 250 μL Western Blotting lysis buffer (see immunoblotting method). The tissue was homogenised on ice and incubated for a further 1 h on ice. The supernatant was then collected by centrifugation (15,000 r.p.m., 10 min, 4°C) in a bench top centrifuge. Supernatants were stored at −80°C until further processing for immunoblotting as above.
References
Fukumura, D. & Jain, R. K. Tumor microenvironment abnormalities: causes, consequences and strategies to normalize. J Cell Biochem 101, 937–949 (2007).
Maxwell, P. H. et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci U S A 94, 8104–8109 (1997).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Semenza, G. L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29, 625–634 (2010).
Wang, G. L., Jiang, B. H., Rue, E. A. & Semenza, G. L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 92, 5510–5514 (1995).
Hu, C. J., Wang, L. Y., Chodosh, L. A., Keith, B. & Simon, M. C. Differential roles of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α in hypoxic gene regulation. Mol Cell Biol 23, 9361–9374 (2003).
Heikkilä, M., Pasanen, A., Kivirikko, K. I. & Myllyharju, J. Roles of the human hypoxia-inducible factor (HIF)-3α variants in the hypoxia response. Cell Mol Life Sci 68, 3885–3901, 10.1007/s00018-011-0679-5 (2011).
Kamura, T. et al. Activation of HIF1α ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci U S A 97, 10430–10435 (2000).
Forsythe, J. A. et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 16, 4604–4613 (1996).
Liu, Y. V. et al. RACK1 competes with HSP90 for binding to HIF-1α and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1α. Mol Cell 25, 207–217 (2007).
Hay, N. & Sonenberg, N. Upstream and downstream of mTOR. Genes Dev 18, 1926–1945 (2004).
Bernardi, R. et al. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature 442, 779–785 (2006).
Semenza, G. L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 24, 97–106 (2009).
Brauch, H. et al. VHL alterations in human clear cell renal cell carcinoma: association with advanced tumor stage and a novel hot spot mutation. Cancer Res 60, 1942–1948 (2000).
Wiesener, M. S. et al. Constitutive activation of hypoxia-inducible genes related to overexpression of hypoxia-inducible factor-1α in clear cell renal carcinomas. Cancer Res 61, 5215–5222 (2001).
Yoshida, M. et al. Somatic von Hippel-Lindau disease gene mutation in clear-cell renal carcinomas associated with end-stage renal disease/acquired cystic disease of the kidney. Genes Chromosomes Cancer 35, 359–364 (2002).
van Houwelingen, K. P. et al. Prevalence of von Hippel-Lindau gene mutations in sporadic renal cell carcinoma: results from The Netherlands cohort study. BMC Cancer 5, 57 (2005).
Banks, R. E. et al. Genetic and epigenetic analysis of von Hippel-Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer Res 66, 2000–2011 (2006).
Nickerson, M. L. et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res 14, 4726–4734, 10.1158/1078-0432.CCR-07-4921 (2008).
Sun, X., Vale, M., Jiang, X., Gupta, R. & Krissansen, G. W. Antisense HIF-1α prevents acquired tumor resistance to angiostatin gene therapy. Cancer Gene Ther 17, 532–540 (2010).
Lee, K. et al. Acriflavine inhibits HIF-1 dimerization, tumor growth and vascularization. Proc Natl Acad Sci U S A 106, 17910–17915 (2009).
Sandau, K. B., Faus, H. G. & Brüne, B. Induction of hypoxia-inducible-factor 1 by nitric oxide is mediated via the PI3K pathway. Biochem Biophys Res Commun 278, 263–267 (2000).
Sutton, K. M. et al. Selective inhibition of MEK1/2 reveals a differential requirement for ERK1/2 signalling in the regulation of HIF-1 in response to hypoxia and IGF-1. Oncogene 26, 3920–3929, 10.1038/sj.onc.1210168 (2007).
Pencreach, E. et al. Marked activity of irinotecan and rapamycin combination toward colon cancer cells in vivo and in vitro is mediated through cooperative modulation of the mammalian target of rapamycin/hypoxia-inducible factor-1α axis. Clin Cancer Res 15, 1297–1307, 10.1158/1078-0432.CCR-08-0889 (2009).
Poon, E., Harris, A. L. & Ashcroft, M. Targeting the hypoxia-inducible factor (HIF) pathway in cancer. Expert Rev Mol Med 11, e26 (2009).
Krieg, M. et al. Up-regulation of hypoxia-inducible factors HIF-1α and HIF-2α under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 19, 5435–5443 (2000).
Raval, R. R. et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol 25, 5675–5686 (2005).
Carroll, V. A. & Ashcroft, M. Role of hypoxia-inducible factor (HIF)-1α versus HIF-2α in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer Res 66, 6264–6270 (2006).
Balasubramanian, S. & Neidle, S. G-quadruplex nucleic acids as therapeutic targets. Curr Opin Chem Biol 13, 345–353 637 (2009).
Rodriguez, R. et al. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat Chem Biol 8, 301–310 (2012).
De Armond, R., Wood, S., Sun, D., Hurley, L. H. & Ebbinghaus, S. W. Evidence for the presence of a guanine quadruplex forming region within a polypurine tract of the hypoxia inducible factor 1α promoter. Biochemistry 44, 16341–16350 (2005).
Balasubramanian, S., Hurley, L. H. & Neidle, S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov 10, 261–275 (2011).
Lombardo, C. M. et al. A novel series of G-quadruplex ligands with selectivity for HIF-expressing osteosarcoma and renal cancer cell lines. Bioorg Med Chem Lett 22, 5984–5988 (2012).
Maxwell, P. H. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275 (1999).
Cockman, M. E. et al. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem 275, 25733–25741 (2000).
Welsh, S. J., Bellamy, W. T., Briehl, M. M. & Powis, G. The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1alpha protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res 62, 5089–5095 (2002).
Pfaffl, M. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45 (2001).
Ohh, M. et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol 2, 423–427 (2000).
Ryan, H. E., Lo, J. & Johnson, R. S. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J 17, 3005–3015 (1998).
Welsh, S. J. et al. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1α and vascular endothelial growth factor formation. Mol Cancer Ther 2, 235–243 (2003).
Xu, Q. et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 24, 5552–5560 (2005).
Isaacs, J. S. et al. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J Biol Chem 277, 29936–29944 (2002).
Bertout, J. A., Patel, S. A. & Simon, M. C. The impact of O2 availability on human cancer. Nat Rev Cancer 8, 967–975 (2008).
Semenza, G. L. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3, 721–732 (2003).
Acknowledgements
Margaret Ashcroft and Patrick Maxwell are thanked for valuable advice, the kind gift of RCC4 cells and pGL3 and derivative vectors. We thank Cancer Research UK for support (Programme grant and Studentship to CL) and the Novartis Institutes for BioMedicalResearch for supplying the cell proliferation data in various renal cancer lines and for the kinase panel data.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: S.J.W., A.G.D., M.F., A.S., S.N. Performed the experiments: S.J.W., A.G.D., C.L., H.V., M.F. Analysed the data: S.J.W., A.G.D., S.N. Contributed reagents, equipment and analysis tools: C.L., A.S., S.N. Wrote the paper: S.J.W., A.G.D., S.N.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Welsh, S., Dale, A., Lombardo, C. et al. Inhibition of the hypoxia-inducible factor pathway by a G-quadruplex binding small molecule. Sci Rep 3, 2799 (2013). https://doi.org/10.1038/srep02799
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02799
This article is cited by
-
G-quadruplexes in MTOR and induction of autophagy
Scientific Reports (2024)
-
The long noncoding RNA LUCAT1 promotes colorectal cancer cell proliferation by antagonizing Nucleolin to regulate MYC expression
Cell Death & Disease (2020)
-
Identification and characterization of G-quadruplex formation within the EP0 promoter of pseudorabies virus
Scientific Reports (2018)
-
Endogenous HIF2A reporter systems for high-throughput functional screening
Scientific Reports (2018)
-
Whole-Transcriptome Profiling of Canine and Human in Vitro Models Exposed to a G-Quadruplex Binding Small Molecule
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
