
Abstract
The mechanisms producing strong coupling between electric and magnetic order in multiferroics are not always well understood, since their microscopic origins can be quite different. Hence, gaining a deeper understanding of magnetoelectric coupling in these materials is the key to their rational design. Here, we use ultrafast optical spectroscopy to show that the influence of magnetic ordering on quantum charge fluctuations via the double-exchange mechanism can govern the interplay between electric polarization and magnetism in the charge-ordered multiferroic LuFe2O4.
Similar content being viewed by others

Introduction
Ferroelectric and ferromagnetic materials possess spontaneous electric and magnetic order, respectively, which can be switched by applied electric and magnetic fields. Multiferroics combine these properties in a single material, providing an avenue for controlling electric polarization with a magnetic field and magnetism with an electric field. These materials have been intensively studied in recent years, both for their fundamental scientific interest as well as their potential applications in a broad range of magnetoelectric devices1,2,3,4.
Recently, the iron-based multiferroic LuFe2O4 has attracted much attention because it exhibits magnetoelectric coupling close to room temperature5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21. The unique layered structure of LuFe2O4 consists of double layers of Fe ions connected in a triangular lattice in the ab-plane (Figure 1(a))14. The average valence of Fe ions is Fe2.5+, with Fe2+ and Fe3+ ions occupying equivalent sites in different layers with equal densities. The corresponding spin values are S = 2 (Fe2+) and S = 5/2 (Fe3+), with the spin structure shown in Figure 1(b)15,16. A simple description based on nearest-neighbour interactions between Fe ions leads to the characterization of this material as a spin and charge frustrated system5,17. Bulk ferroelectricity was observed below the charge ordering temperature, TCO ~ 320 K, resulting in a spontaneous electric polarization that further increased upon the appearance of ferrimagnetic spin order below the Neel temperature, TN ~ 240 K5. Ferroelectricity in each bilayer is thus induced by electronic charge ordering, although the stacking of adjacent bilayers (i.e., in an antiferroelectric or ferroelectric arrangement) is still controversial7,8,11,12,15,18. Regardless, in each bilayer, the electric polarization P is coupled to the magnetic degree of freedom in LuFe2O4, but a comprehensive understanding of the mechanism underlying this magnetoelectric coupling has eluded researchers to date. Knowledge of this mechanism could potentially allow researchers to optimize both the strength of this coupling and its operating temperature to address the general goal of developing multiferroic materials with strong magnetoelectric coupling at room temperature.

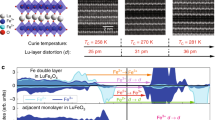
Charge and spin ordering in LuFe2O4 above and below TN.
(a) For T > TN, charge ordering results in a finite polarization P. The top layer is displaced from the bottom layer by an angle shown by the black straight dashed line3; this is also shown clearly in Figure 3. Quantum fluctuations between Fe2+ and Fe3+ ions (depicted by blue dashed lines) can reduce P by delocalizing charges, with an effective matrix element for hopping between two sites given by t12 = t/2. Red arrows show the possible charge transfer routes between Fe2+ and Fe3+, as defined in the text. (b) For T < TN, ferrimagnetic spin ordering increases P by decreasing the average hopping through the double exchange mechanism; t12 = t when spins at both sites are ferromagnetically aligned (blue dashed lines) and t12 = 0 when both spins are antiferromagnetically aligned (red dashed lines).
Theoretical studies have linked the magnetoelectric coupling in LuFe2O4 to both thermal19 and quantum20,21 charge fluctuations. In general, magnetic ordering can modify the effective hopping amplitude between two ions via the well-known double-exchange mechanism20,21,22,23, in which hopping is governed by the angle between the two core spins, as shown in Fig. 1(b). This change in the hopping amplitude will necessarily affect the quantum fluctuations of any charge ordered state of electronic origin. The corresponding change in charge ordering will necessarily modify P. In other words, if electronic charge ordering leads to a net electric polarization, the value of P should be modified by the presence of magnetic ordering (Fig. 1(b)). Moreover, if the magnetic ordering reduces (on average) the effective hopping amplitude, the corresponding suppression of quantum charge fluctuations leads to an increase of P below TN. Since the same mechanism should affect optically induced charge fluctuations, we use femtosecond optical pump-probe spectroscopy, which has been extensively used to shed light on the properties of correlated electron materials24,25,26,27,28,29,30,31, to directly photoexcite and probe the Fe2+→Fe3+ charge transfer channel in LuFe2O4. Then, by varying the temperature T above and below TN, we can shed light on the role of these fluctuations in governing the coupling between spin and charge order in a single LuFe2O4 bilayer, regardless of whether the bilayers are stacked ferroelectrically or antiferroelectrically. We find that the interlayer hopping matrix element describing these fluctuations depends strongly on their local core spin alignment via the double-exchange mechanism, making charge delocalization (in real space, as shown in Fig. 1(b)) and hence the electric polarization extremely sensitive to the spin structure evolution over a broad temperature range. Therefore, although magnetoelectric coupling in various multiferroic materials has been studied using many different techniques32, to the best of our knowledge, this is the first experimental evidence of magnetoelectric coupling mediated by the double-exchange mechanism in an insulator.
Results
We begin by developing a model for electronic hopping between two atomic sites, governed by the double exchange mechanism, which shows that the transition rate (i.e., charge transfer rate) between the ground and the excited state induced by an external driving electromagnetic field is proportional to the effective hopping matrix, tij2, according to the Fermi Golden rule33, as described in Methods. Furthermore, the amount of delocalized charge δq is proportional to tij2,  , where Δ is the energy difference between the ground and excited states. This simple observation establishes our ability to probe quantum charge fluctuations between two atomic sites using optical spectroscopy (see Methods for more detail). In LuFe2O4, these fluctuations are due to charge transfer between Fe2+ and Fe3+ ions (as revealed by optical spectroscopy34 and band structure calculations19). We consider four different charge transfer channels in the bilayer crystal structure of LuFe2O4: interlayer charge transfer from the Fe2+ rich bottom layer to the Fe3+ rich top layer (E↑) or from the top to the bottom layer (E↓) and intralayer charge transfer within the top layer (Et→) and within the bottom layer (Eb→), as shown in Fig. 1(a). We can gain insight on the relative energies of these different charge transfer channels by considering the Coulomb energy between Fe ions in the Hamiltonian,
, where Δ is the energy difference between the ground and excited states. This simple observation establishes our ability to probe quantum charge fluctuations between two atomic sites using optical spectroscopy (see Methods for more detail). In LuFe2O4, these fluctuations are due to charge transfer between Fe2+ and Fe3+ ions (as revealed by optical spectroscopy34 and band structure calculations19). We consider four different charge transfer channels in the bilayer crystal structure of LuFe2O4: interlayer charge transfer from the Fe2+ rich bottom layer to the Fe3+ rich top layer (E↑) or from the top to the bottom layer (E↓) and intralayer charge transfer within the top layer (Et→) and within the bottom layer (Eb→), as shown in Fig. 1(a). We can gain insight on the relative energies of these different charge transfer channels by considering the Coulomb energy between Fe ions in the Hamiltonian,

where the pseudospin operators  and
and  are 1/2 or −1/2 for Fe3+ or Fe2+, respectively and ε0, εr and rij are the permittivity of free space, the relative permittivity and the distance between sites i and j, respectively. Considering only the largest three interaction terms, we find that E↑ has the lowest excitation energy, Et→ and Eb→ have intermediate excitation energies and E↓ has the highest excitation energy. The interlayer transitions can be distinguished by noting that the bottom layer is rich in Fe2+ while the top layer is rich in Fe3+. Therefore, it is clear that if the top layer has a positive charge density σ > 0 per unit area, the bottom layer must have the opposite charge density, −σ, to ensure charge neutrality. E↓ increases with σ while E↑ decreases, so it is reasonable to assume that E↓ ≫ E↑ in LuFe2O4. We can also distinguish the intralayer transitions by noting that the configuration of in-plane oxygen ions around Fe ions in both layers leads to a higher in-plane charge transfer excitation energy for Eb→ than that of Et→. The optical conductivity measurements described in ref. 34 show two distinct charge transfer excitation channels at ~1.1 eV and ~1.5 eV, which should thus correspond to E↑ and Et→, respectively. To further confirm this, we performed angle-dependent reflectivity measurements with a p polarized laser beam at different incident angles (25 and 45 degrees), which allowed us to extract the imaginary part of the refractive index (Figure 2(a)). Our data clearly shows two peaks at ~1.1 and 1.5 eV, in good agreement with the data from ref. 34, which is also plotted for comparison. It is worth noting that our experiments and those of ref. 34 were performed in different setups on samples grown by different groups; the close agreement between the data sets thus gives additional confidence in our results.
are 1/2 or −1/2 for Fe3+ or Fe2+, respectively and ε0, εr and rij are the permittivity of free space, the relative permittivity and the distance between sites i and j, respectively. Considering only the largest three interaction terms, we find that E↑ has the lowest excitation energy, Et→ and Eb→ have intermediate excitation energies and E↓ has the highest excitation energy. The interlayer transitions can be distinguished by noting that the bottom layer is rich in Fe2+ while the top layer is rich in Fe3+. Therefore, it is clear that if the top layer has a positive charge density σ > 0 per unit area, the bottom layer must have the opposite charge density, −σ, to ensure charge neutrality. E↓ increases with σ while E↑ decreases, so it is reasonable to assume that E↓ ≫ E↑ in LuFe2O4. We can also distinguish the intralayer transitions by noting that the configuration of in-plane oxygen ions around Fe ions in both layers leads to a higher in-plane charge transfer excitation energy for Eb→ than that of Et→. The optical conductivity measurements described in ref. 34 show two distinct charge transfer excitation channels at ~1.1 eV and ~1.5 eV, which should thus correspond to E↑ and Et→, respectively. To further confirm this, we performed angle-dependent reflectivity measurements with a p polarized laser beam at different incident angles (25 and 45 degrees), which allowed us to extract the imaginary part of the refractive index (Figure 2(a)). Our data clearly shows two peaks at ~1.1 and 1.5 eV, in good agreement with the data from ref. 34, which is also plotted for comparison. It is worth noting that our experiments and those of ref. 34 were performed in different setups on samples grown by different groups; the close agreement between the data sets thus gives additional confidence in our results.

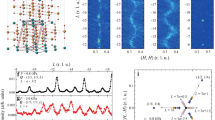
Angle-dependent reflectivity measurements in LuFe2O4.
(a) Imaginary part of the refractive index for p polarized excitation at incident angles of 25 and 45 degrees. (b) The data from (a), normalized to its value at 1.1 eV in order to compare the relative peak intensities at 1.5 eV.
Figure 2 supports our assignment of the two observed peaks to the energies E↑ (1.1 eV) and Et→ (1.5 eV), due to inter- and intra-layer charge transfer, respectively, as follows: As we increase the incident angle in our reflectivity measurements, we expect the absorption at E↑ to increase and Et→ to decrease for p polarized light. This can be seen more clearly in Fig. 2(b), in which we normalized the data of Fig. 2(a) to the peak at 1.1 eV to facilitate the comparison of both peaks. This shows that the relative strength of the 1.5 eV peak, as compared to the 1.1 eV interlayer charge transfer peak, becomes weaker as we increase the incident angle, supporting our assignment of these two peaks. It is worth noting that both in our measurements and in the data of ref. 34, no spectral signatures corresponding to Eb→ and E↓ were observed. This is likely because there are many different possible transitions that overlap at higher energies, which obscure the peaks corresponding to Eb→ and E↓. Therefore, we used photon energies of 1.1 (E↑) and 1.5 (Et→) eV in our experiments to examine inter- and intralayer quantum charge fluctuations in LuFe2O4.
We propose that magnetic order and charge fluctuations in LuFe2O4 are linked through the double exchange mechanism20,21, which leads to an effective hopping matrix element tij (see Methods) between the ions i and j that is determined by the angle θij between the spins Si and Sj : tij = tcos(θij/2)23. Within an Ising spin model, the hopping matrix element tij will be 0 (if the Fe2+ and Fe3+ core spins are antiparallel) or t (if they are parallel). Thermal fluctuations prevent any preferred spin orientation in the paramagnetic phase (T > TN), so tij will be given by its average value of t/2 (Fig. 1(a)) (similarly, tij2 = 0 or t2, respectively and thus its average value will be t2/2 for this transition). However, in the magnetically ordered state (T < TN), tij = t if the nearest neighbour spins are aligned ferromagnetically (FM), while tij = 0 if they are antiferromagnetically (AFM) aligned (Fig. 1(b) and Figure 3). This can then be applied to the four charge transfer channels discussed above (considering electron hopping from a given Fe2+ ion to its nearest neighbour Fe3+ ions) to find the total steady-state absorption (proportional to tij2 (see Methods)) for each transition above and below TN. We find that the development of magnetic order does not affect the total tij2 for E↑ and Et→. This can straightforwardly be seen by considering the E↑ transition (Fig. 1(a)), where the spins do not have a preferred direction above TN. Because there are two possible charge transfer channels for E↑ (Fig. 1(a)), we obtain a total transition amplitude of t2/2 × 2 = t2 above TN. Below TN, there is only one possible transition with an amplitude of t2 (Fig. 1(b)), so the transition amplitudes above and below TN are the same. In the same manner, if we consider the Et→ transition above TN (Fig. 1(a)), there are six charge transfer channels, giving a transition amplitude of t2/2 × 6 = 3t2. Below TN, there are only three possible transitions, each with amplitude t2, so the transition amplitudes above and below TN are the same, as for the interlayer transition.

Possible interlayer and intralayer charge transfer transitions for T < TN.
Both figures show the spin structure of LuFe2O4 for T < TN. Big and small solid circles correspond to atoms in the top and bottom layers, respectively. The black arrows show the local spins along the c axis at 220 K, from refs. 15 and 16. Red solid and broken arrows represent allowed and forbidden interlayer (a) and intralayer (b) charge transfer channels, respectively, when considering the double exchange mechanism. After 1.1 eV excitation, one electron from an Fe2+ ion in the bottom layer moves to an Fe3+ ion in the top layer, while after 1.5 eV excitation, one electron from an Fe2+ site in the top layer moves to an Fe3+ site in the same layer.
This argument thus verifies that there is no change in the steady-state absorption for both transitions examined here (Et→ and E↑) between T > TN and T < TN. This is also clear from the data in ref. 34, which shows that the absorption for the E↑ and Et→ transitions does not significantly change with temperature. In contrast, the total tij2 is reduced through the double exchange mechanism for Eb→ and E↓ below TN; therefore, as described above in the introduction, the corresponding suppression of quantum charge fluctuations should increase P. A simple test of the influence of the double exchange mechanism on magnetoelectric coupling in LuFe2O4 would thus be to track the expected changes in the steady-state optical absorption at Eb→ and E↓ as the temperature T is varied across TN. However, the conductivity data in ref. 34 does not show any distinct spectral signature that could be attributed to these charge transfer energies, likely due to the presence of many overlapping transitions in this energy range as described earlier.
One can circumvent this limitation by performing a non-equilibrium experiment; i.e., photoexciting the known E↑ and Et→ transitions and examining the resulting changes in the optical absorption at the E↑ transition (which is proportional to P). Photoexcitation changes the charge configuration, which in turn changes both the total tij2 and the E↑ charge transfer energy through equation (1), leading to a transient change in the reflectivity that can be measured in our experiments (see Methods for more detail). The development of magnetic order below TN can then further modify the absorption at E↑ through the double exchange mechanism in the same manner as in the steady state, potentially causing an additional change in the photoinduced reflectivity. In short, our ultrafast optical experiments (described in more detail in Methods) allow us to examine the effect of photoexciting either intralayer or interlayer charge transfer transitions (in effect externally driving charge fluctuations) on the interlayer charge transfer energy as a function of temperature. Tuning the temperature above and below TN then allows us to determine the effect of magnetic ordering on charge fluctuations. In this way, we can test if these fluctuations are indeed responsible for the magnetoelectric coupling measured in LuFe2O4.
Figure 4(a) shows the temporal profile of the normalized photoinduced reflectivity change, ΔR/R(t), in LuFe2O4 at several different temperatures for a degenerate 1.1 eV pump-probe measurement. Immediately after photoexcitation, ΔR/R decreases to its minimum value within <0.5 picoseconds (ps) (Figure 4(b)) and returns to equilibrium while exhibiting coherent acoustic phonon oscillations with a period of ~40 ps (Fig. 4(a)). These coherent phonons are generated by the dynamic stress on the sample induced by absorption of the pump pulses and are commonly observed in ultrafast optical experiments on correlated electron materials30, as well as non-correlated materials described by a simple band structure35. In this paper, we will focus on the variation of the maximum amplitude of the transient reflectivity signal (ΔR/Rmax) with temperature, which gives insight into the influence of intra- and interlayer charge fluctuations on magnetoelectric coupling in LuFe2O4.

Temperature-dependent transient reflectivity change after 1.1 eV photoexcitation.
(a) Temperature-dependent transient reflectivity change after photoexciting and probing the Fe2+ → Fe3+ interlayer charge transfer excitation at 1.1 eV. The open circles are the experimentally obtained data points and the solid lines are the results of fitting the data with exponential and oscillating terms. (b) The data from (a) at early times. (c) The amplitude of the negative peak as a function of temperature. The red vertical line shows the magnetic transition temperature, TN ~ 240 K and the blue horizontal line is parallel to the x axis, which indicates that there is no significant variation in ΔR/Rmax (1.1 eV) across TN. We note that the high signal-to-noise ratio of our experiment resulted in error bars that are comparable to the size of the data points and are therefore not shown.
Figure 4(c) depicts ΔR/Rmax (1.1 eV) after 1.1 eV photoexcitation as a function of temperature near TN. As described above, photoexcitation changes the charge distribution in LuFe2O4, altering the energy required to transfer a charge from the bottom to the top layer (E↑) (and thus the absorption/reflectivity probed at 1.1 eV) (Figure 3 (a)). We calculated ΔE↑, the difference in the interlayer charge transfer energy before and after photoexcitation, for T < TN and T > TN, using equation (1) (and using εr = 2 from ref. 34) (see Methods). This calculation reveals that there is no change in the ΔR/Rmax signal as T is varied across TN, which is consistent with our experimental observation (Fig. 4(c)); fundamentally, although the specific allowed charge transfer transitions after photoexcitation do change across TN due to the double exchange mechanism (Fig. 3(a)), the net effect of magnetic ordering on the resulting absorption change is very small and thus undetectable in our experiment. The negative sign of the signal is also expected since photoexcitation reduces the absorption at 1.1 eV.
Figure 5(a) shows the transient reflectivity change for the 1.1 eV interlayer transition after photoexciting the intralayer (Et→) charge transfer channel at 1.5 eV. The time-dependent dynamics are similar to those observed after photoexciting E↑, but the variation of ΔR/Rmax with temperature is quite different (Figure 5(b)); in particular, a significant increase in the amplitude is clearly observed as the temperature rises above TN. As described above, the steady state absorption for the 1.1 eV (E↑) interlayer transition does not change across TN; however, the probe absorption at this transition can change after 1.5 eV photoexcitation as T is varied across TN. To understand this, we calculated the effect of the photoinduced intralayer charge transfer at 1.5 eV on E↑ in the same manner as described above for E↑ (see Methods). Our calculation shows that the ratio of the maximum photoinduced change in reflectivity between T < TN and T > TN is ~0.9, which agrees very well with our experimental results (~0.87) (Fig. 5(b)).

Temperature-dependent transient reflectivity change after 1.5 eV photoexcitation.
(a) Transient reflectivity change at 1.1 eV for temperatures of 200 and 300 K after photoexciting the Fe2+ → Fe3+ intralayer charge transfer excitation at 1.5 eV. (b) Peak amplitude of the transient reflectivity change near TN after 1.5 eV excitation.
Discussion
From this experimental observation, we deduce that the ferrimagnetic order influences the fluctuations of the charge ordered state that is responsible for the electric polarization in LuFe2O4 through the double exchange interaction. This result indicates that the interplay between charge fluctuations and magnetic ordering can result in magnetoelectric coupling at the Neel temperature. Finally, it is worth noting that this mechanism will generate an electric polarization in each bilayer, regardless of whether the ground state consists of layers stacked with ferroelectric or antiferroelectric order.
In summary, we used femtosecond optical pump-probe spectroscopy to investigate the role of the double exchange mechanism in the magnetoelectric coupling observed in LuFe2O4. Our experiments revealed that optically induced charge fluctuations are affected by magnetic order in a manner that is consistent with this mechanism. Importantly, this result opens an alternative route for finding strong magnetoelectric effects: charge ordering in transition metal oxides can naturally lead to electric polarization that is coupled to the magnetic degree of freedom via the double-exchange interaction.
Methods
Experimental setup and sample preparation
Our femtosecond optical pump-probe spectroscopy system is based on a 75 femtosecond (fs), 250 kHz repetition rate Ti:sapphire amplified laser system operating at 800 nm (~1.5 eV) and seeding an optical parametric amplifier (OPA) that allows us to tune the photon energy. A delay line allows us to vary the optical path difference between the pump (200 μm diameter) and probe (100 μm diameter) beams, which are then focused to the same spot on the sample. Temperature-dependent transient reflectivity changes were obtained in reflection with cross-polarized pump and probe beams in a >10:1 power ratio (the pump fluence is 76 μJ/cm2, which photoexcites ~0.007 electrons/unit cell), incident at an angle of less than 10° to the hexagonal c axis of the crystal. The probe photon energy was 1.1 eV in all experiments (examining E↑) and the pump photon energies were 1.1 and 1.5 eV (photoexciting E↑ and Et→, respectively). We emphasize that the interlayer Fe-Fe transition dipole is not oriented perpendicular to the surface (and thus not aligned with the c-axis since Fe ions in the top layer are not located above Fe ions in the bottom layer, but instead above O ions) (Figs. 1 and 3)3,6,13,15,16, so our near normal incident 1.1 eV pulses can sensitively photoexcite and probe changes in its absorption. It is also worth mentioning that pump fluence-dependent measurements revealed that only the amplitude of the ΔR/R signal changes linearly with fluence, with no changes in the measured dynamics, for both 1.1 and 1.5 eV excitation. Furthermore, at the fluence used here, the maximum temperature increase due to laser heating is <8 K, which should not significantly affect the measured dynamics and the sample completely recovers in the 4 μs time interval between amplifier pulses. Finally, the LuFe2O4 single crystal used in this study was grown by the floating zone method as described in ref. 15, with its surface normal to the c-axis.
Theoretical background
Quantum charge fluctuations originate from hopping of an electron between two spatially separated potential minima. When an electron is localized in one potential well, the system becomes electrically polarized20,21. If we consider two energetically non-degenerate states localized at two different lattice sites and include the double exchange interaction in the system, the Hamiltonian can be expressed as

where εi is the atomic energy,  and Ci are creation and annihilation operators at the ith site (i = 1, 2) and tij = tji is a effective hopping matrix element accounting for the hopping between sites. This matrix element is governed by the double exchange mechanism, which relates the probability of an electron hopping between two atoms to the angle θij between the local core spins Si and Sj22,23. This mechanism has most frequently been used to explain the metallic properties of colossal magnetoresistive manganites22, but also applies here to LuFe2O4, since the ferrimagnetic spin order existing below TN influences electron hopping (and therefore charge fluctuations of the charge-ordered state), which can in turn change the dielectric properties of the system. In other words, since the electronic charge is never completely localized in insulators, the degree of delocalization depends on the effective hopping amplitude given by the double exchange mechanism. Since magnetic ordering suppresses this hopping amplitude for the Eb→ and E↓ transitions, we expect electrons in LuFe2O4 to be more localized, stabilizing charge order.
and Ci are creation and annihilation operators at the ith site (i = 1, 2) and tij = tji is a effective hopping matrix element accounting for the hopping between sites. This matrix element is governed by the double exchange mechanism, which relates the probability of an electron hopping between two atoms to the angle θij between the local core spins Si and Sj22,23. This mechanism has most frequently been used to explain the metallic properties of colossal magnetoresistive manganites22, but also applies here to LuFe2O4, since the ferrimagnetic spin order existing below TN influences electron hopping (and therefore charge fluctuations of the charge-ordered state), which can in turn change the dielectric properties of the system. In other words, since the electronic charge is never completely localized in insulators, the degree of delocalization depends on the effective hopping amplitude given by the double exchange mechanism. Since magnetic ordering suppresses this hopping amplitude for the Eb→ and E↓ transitions, we expect electrons in LuFe2O4 to be more localized, stabilizing charge order.
For small tij values (tij ≪ Δ, where ε2 − ε1 ≡ Δ and t12 = t21) and ε2 > ε1, most of the charge will be localized at site 1, with a small fraction  of delocalized charge remaining at site 2. Because the electric polarization P is proportional to the difference of charge densities between sites, ρ2 − ρ1 (where ρ2 and ρ1 are the electron densities at sites 2 and 1, respectively), any change in the delocalized charge at site 2 causes a change in P.
of delocalized charge remaining at site 2. Because the electric polarization P is proportional to the difference of charge densities between sites, ρ2 − ρ1 (where ρ2 and ρ1 are the electron densities at sites 2 and 1, respectively), any change in the delocalized charge at site 2 causes a change in P.
When an external electromagnetic field (E0cosωt) is applied to the system, it will introduce a small perturbation H1 = exE0cosωt into the Hamiltonian, inducing a site-to-site transition (where e is the electron charge and x is the distance between the two sites). We can use Fermi's golden rule33 to calculate the probability of transitions between both sites (corresponding to quantum charge fluctuations), which is found to be proportional to the delocalized charge on site 2, δq, through  (where
(where  and
and  are the ground and excited states of H0, respectively). Note that the transition rate is proportional to the extent of charge delocalization in the ground state of the system. Since the photoinduced change in reflectivity at the absorption peak is proportional to changes in the absorption under the conditions of our experiment31, which, in turn, is proportional to
are the ground and excited states of H0, respectively). Note that the transition rate is proportional to the extent of charge delocalization in the ground state of the system. Since the photoinduced change in reflectivity at the absorption peak is proportional to changes in the absorption under the conditions of our experiment31, which, in turn, is proportional to  and
and  , we can relate our transient reflectivity measurements to the amount of delocalized charge and thus the polarization P. This then establishes that we can use our ultrafast optical measurements to reliably photoexcite and probe quantum charge fluctuations in LuFe2O4.
, we can relate our transient reflectivity measurements to the amount of delocalized charge and thus the polarization P. This then establishes that we can use our ultrafast optical measurements to reliably photoexcite and probe quantum charge fluctuations in LuFe2O4.
We calculated ΔE↑, i.e. the pump-induced difference in the interlayer charge transfer energy, using equation (1) and only considering four ions: two excited by the pump and two examined by the probe after photoexcitation (Figure 3 in our manuscript). This was done by exchanging a Fe2+ and a Fe3+ ion either between the bottom and top layers (corresponding to absorption of a 1.1 eV pump photon) (Figure 3(a)) or between two sites in the top layer (corresponding to absorption of a 1.5 eV pump photon) (Figure 3(b)). We then obtained ΔE↑ based on the new Coulomb energy after exchanging Fe2+ and Fe3+ ions. This was then used to calculate the change in absorption as described above, from which we calculated the variation of ΔR/Rmax with temperature after both 1.1 eV and 1.5 eV photoexcitation for comparison to our experimental data. It is worth noting that if either of the ions that absorb a pump photon is involved in the subsequent absorption of a probe photon, we find that the resulting ΔE↑ is much larger than the probe bandwidth (~13 meV) and thus does not contribute to the observed absorption change. Finally, including more than 47 Fe2+ electrons in this calculation resulted in an insignificant reflectivity change, since there is almost no change in E↑ due to Fe2+ ions far from the Fe2+ and Fe3+ sites that participate in the photoinduced transition.
References
Eerenstein, W., Mathur, N. D. & Scott, J. F. Multiferroic and magnetoelectric materials. Nature 442, 759–765 (2006).
Chu, Y.-H., Martin, L. W., Holcomb, M. B. & Ramesh, R. Controlling magnetism with multiferroics. Mater. Today 10, 16–23 (2007).
Cheong, S.-W. & Mostovoy, M. Multiferroics: a magnetic twist for ferroelectricity. Nature. Mater. 6, 13–20 (2007).
Khomskii, D. Classifying multiferroics: Mechanisms and effects. Physics 2, 20 (2009).
Ikeda, N. et al. Ferroelectricity from iron valence ordering in the charge-frustrated system LuFe2O4 . Nature 436, 1136–1138 (2005).
Subramanian, M. A. et al. Giant room-temperature magnetodielectric response in the electronic ferroelectric LuFe2O4 . Adv. Mater. 18, 1737–1739 (2006).
Niermann, D., Waschkowski, F., de Groot, J., Angst, M. & Hemberger Dielectric Properties of Charge-Ordered LuFe2O4 Revisited: The Apparent Influence of Contacts. Phys. Rev. Lett. 109, 016405 (2012).
de Groot, J. et al. Competing Ferri- and Antiferromagnetic Phases in Geometrically Frustrated LuFe2O4 . Phys. Rev. Lett. 108, 037206 (2012).
Lee, C., Kim, J.-Y. & Cheong, S. W. Infrared optical response of LuFe2O4 under dc electric field. Phys. Rev. B 85, 014303 (2012).
Mulders, A. M., Bartkowiak, M., Hester, J. R., Pomjakushina, E. & Conder, K. Ferroelectric charge order stabilized by antiferromagnetism in multiferroic LuFe2O4 . Phys. Rev. B 84, 140403 (R) (2011).
Xu, X. S. et al. Lattice dynamical probe of charge order and antipolar bilayer stacking in LuFe2O4 . Phys. Rev. B 82, 014304 (2010).
Vitucci, F. M. et al. Infrared study of the charge-ordered multiferroic LuFe2O4 . Phys. Rev. B 81, 195121 (2010).
Ishihara, S. Electric Ferroelecricity and Frustration. J. Phys. Soc. Jpn. 79, 011010 (2010).
Iida, J. et al. Magnetization and spin correlation of two-dimensional triangular antiferromagnet LuFe2O4 . J. Phys. Soc. Jpn. 62, 1723–1735 (1993).
Ko, K.-T. et al. Electronic origin of giant magnetic anisotropy in multiferroic LuFe2O4 . Phys. Rev. Lett. 103, 207202 (2009).
Xiang, H. J., Kan, E. J., Wei, S.-H., Whangbo, M.-H. & Yang, J. Origin of the Ising ferrimagnetism and spin-charge coupling in LuFe2O4 . Phys. Rev. B 80, 132408 (2009).
Yamada, Y., Nohdo, S. & Ikeda, N. Incommensurate charge ordering in charge-frustrated LuFe2O4 system. J. Phys. Soc. Jpn. 66, 3733–3736 (1997).
Angst, M. et al. Charge order in LuFe2O4: Antiferroelectric ground state and coupling to magnetism. Phys. Rev. Lett. 101, 227601 (2008).
Xiang, H. J. & Whangbo, M.-H. Charge order and the origin of giant magnetocapacitance in LuFe2O4 . Phys. Rev. Lett. 98, 246403 (2007).
Watanabe, T. & Ishihara, S. Quantum fluctuation and geometrical frustration effects on electric polarization. J. Phys. Soc. Jpn. 78, 113702 (2009).
Watanabe, T. & Ishihara, S. Quantum dielectric fluctuation in electronic ferroelectricity studied by variational Monte-Carlo method. J. Phys. Soc. Jpn. 79, 114714 (2010).
Tokura, Y. Colossal Magnetoresistive Oxides. (Gordon and Breach Science, Amsterdam, 2000).
Khomskii, D. Electronic structure, exchange and magnetism in oxides. In Lecture Notes on Physics, ed. Thornton M. J. & Ziese M. (Springer-Verlag, Berlin, 2001).
Eichberger, M. et al. Snapshot of cooperative atomic motions in the optical suppression of charge density waves. Nature 468, 799–802 (2010).
Möhr-Vorobeva, E. et al. Nonthermal melting of a charge density waves in TiSe2 . Phys. Rev. Lett. 107, 036403 (2011).
Prasankumar, R. P. et al. Coupled Charge-Spin Dynamics of the Magnetoresistive Pyrochlore Tl2Mn2O7 Probed Using Ultrafast Midinfrared Spectroscopy. Phys. Rev. Lett. 95, 267404 (2005).
Talbayev, D. et al. Detection of Coherent Magnons via Ultrafast Pump-Probe Reflectance Spectroscopy in Multiferroic Ba0.6Sr1.4Zn2Fe12O22 . Phys. Rev. Lett. 101, 97603 (2008).
Först, M. et al. Nonlinear phononics as an ultrafast route to lattice control. Nature Phys. 7, 854–856 (2011).
Matsubara, M., Kaneko, Y., He, J.-P., Okamoto, H. & Tokura, Y. Ultrafast polarization and magnetization response of multiferroic GaFeO3 using time-resolved nonlinear optical techniques. Phys. Rev. B 79, 140441(R) (2009).
Ren, Y. H. et al. Generation and detection of coherent longitudinal acoustic phonons in the La0.67Sr0.33MnO3 thin films by femtosecond light pulses. Appl. Phys. Lett. 90, 251918 (2007).
Gong, T. et al. Femtosecond optical response of Y-Ba-Cu-O films: The dependence on optical frequency, excitation intensity and electric current. Phys. Rev. B 47, 14495–14502 (1993).
van den Brink & Khomskii Daniel, I. Multiferroicity due to charge ordering. J. Phys.: Condens. Matter 20, 434217 (2008).
Liboff, R. L. Introductory Quantum Mechanics. (Addison Wesley, Reading, 1992).
Xu, X. S. et al. Charge order, dynamics and magnetostructural transition in multiferroic LuFe2O4 . Phys. Rev. Lett. 101, 227602 (2008).
Liu, R. et al. Femtosecond pump-probe spectroscopy of propagating coherent acoustic phonons in InxGa1−xN/GaN heterostructures. Phys. Rev. B 72, 195335 (2005).
Acknowledgements
This work was performed under the auspices of the Department of Energy, Office of Basic Energy Sciences, Division of Material Sciences. Los Alamos National Laboratory, an affirmative action equal opportunity employer, is operated by Los Alamos National Security, LLC, for the National Nuclear Security administration of the U.S. Department of Energy under contract no. DE-AC52-06NA25396. The work at Rutgers University was supported by the DOE under Grant No. DE-FG02-07ER46382.
Author information
Authors and Affiliations
Contributions
J.L. and D.T. performed the ultrafast optical experiments, with assistance from D.A.Y. and guidance from D.A.Y., A.J.T. and R.P.P., C.L.Z. and S.-W.C. grew the LuFe2O4 samples. X.S.X. provided optical conductivity data. J.L. analyzed the experimental data and J.L., S.A.T., C.D.B., A.J.T. and R.P.P. worked together to interpret and understand the data, with additional input from X.S.X. and S.-W.C., J.L. and R.P.P. wrote the majority of the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Lee, J., Trugman, S., Batista, C. et al. Probing the Interplay between Quantum Charge Fluctuations and Magnetic Ordering in LuFe2O4. Sci Rep 3, 2654 (2013). https://doi.org/10.1038/srep02654
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02654
This article is cited by
-
The evolution of multiferroics
Nature Reviews Materials (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
