
Abstract
Graphene has a unique atom-thick two-dimensional structure and excellent properties, making it attractive for a variety of electrochemical applications, including electrosynthesis, electrochemical sensors or electrocatalysis and energy conversion and storage. However, the electrochemistry of single-layer graphene has not yet been well understood, possibly due to the technical difficulties in handling individual graphene sheet. Here, we report the electrochemical behavior at single-layer graphene-based electrodes, comparing the basal plane of graphene to its edge. The graphene edge showed 4 orders of magnitude higher specific capacitance, much faster electron transfer rate and stronger electrocatalytic activity than those of graphene basal plane. A convergent diffusion effect was observed at the sub-nanometer thick graphene edge-electrode to accelerate the electrochemical reactions. Coupling with the high conductivity of a high-quality graphene basal plane, graphene edge is an ideal electrode for electrocatalysis and for the storage of capacitive charges.
Similar content being viewed by others

Introduction
The atom-thick two-dimensional structure, high electrical conductivity, large specific surface area and excellent electrochemical stability make graphene a unique electrode material1,2,3,4,5,6,7. At the molecular level, a single-layer graphene sheet has two different structural regions: (1) the basal plane, consisting of two-dimensional conjugated sp2 carbon atoms; (2) the edge, making of one-atom thick defective graphitic line of carbon atoms with dandling bonds and various capping moieties (e.g., hydrogen, hydroxyl, carbonyl and carboxyl groups)8,9. It is believed that the electrochemical behavior at graphitic basal plane is different from that at its edge, while the conclusions of previous studies are confusion10,11. For example, it was reported that the surface defects of highly oriented pyrolytic graphite (HOPG) in the form of exposed edge-plane nanobands exhibited an electron transfer-rate constant (k) about 7 orders of magnitude higher than that at its basal plane12 and the k value of a defect-free HOPG was found to be around zero8,9. In contrast, several groups also claimed that the basal plane of HOPG was not totally inert and electron transfer occurred not only at the step edges of HOPG but also at the pristine graphitic basal planes13,14. Carbon nanotube (CNT), a sibling of graphene, is a one-dimensional carbon electrode. It was revealed that the open ends of CNTs had an accelerated charge-transfer rate and an enhanced electrocatalytic activity than their side walls15,16,17,18. The side walls of CNTs were suggested to have electrochemical properties similar to those of the basal plane of HOPG16,19. On the other hand, several papers also reported that both the ends and side walls of CNTs were electrochemically active18 and their relative electrochemical activities varied with different electrochemical probes because of the difference in their reaction mechanisms20. The edge of a single-layer graphene sheet is different from the surface edge-plane of HOPG and the end of CNT, mainly due to its linear atom-thick structure. Here, we report the electrochemistry at either edge- or basal plane-based graphene electrode. It was found that the graphene edge has much larger specific capacitance, faster electron transfer rate and stronger electrocatalytic activity than those of its basal plane.
Results
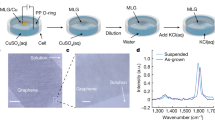
High-quality single-layer graphene sheets with lateral dimensions of several centimeter squares were synthesized by chemical vapor deposition (CVD) on a Cu substrate (Methods). In order to perform the edge- and basal plane-selective electrochemistry for a single-layer graphene sheet, we coated the graphene sheet region-specifically with a non-conducting pinhole-free thin film (e.g., epoxy resin, ER). Figure S1 illustrates the procedures of fabricating the graphene electrodes. As shown in this figure, ER was selectively coated around the graphene sheet to leave a square window for fabricating the plane-electrode. The exposed Cu substrate underneath the window was then etched with an aqueous solution of FeCl3 and followed by rinsing with de-ionized water. For fabricating the edge-electrode, the whole graphene sheet was encapsulated in ER and then vertically cut to expose its cross-section. Two corner regions of the newly-formed graphene cross-section were further coated with ER to leave only 1–2 mm uncovered edge for minimizing the possibility of cracking between the graphene layer and resin coating during the electrochemical processes (Figure S2a). Figure 1a schematically shows the geometrical configurations of the resulting edge- and plane-electrodes.

The configurations of plane- and edge-electrodes and the structure of a CVD graphene sheet.
(a) Schematic illustration of the edge-based (left) and basal plane-based (right) electrodes. (b) Raman spectrum of a monolayer CVD graphene sheet on a SiO2/Si substrate. (c) HR-TEM image of a monolayer CVD graphene sheet suspended over a micro grid; inset: a SAED pattern.
Prior to the electrochemical measurements, the quality and uniformity of the CVD grown single-layer graphene sheet were characterized by Raman spectroscopy and high-resolution transmission electron microscopy (HR-TEM, Figure 1b and 1c). Figure 1b has a G-band and a 2D-band at 1585 and 2690 cm−1, respectively. The weak band located at 1350 cm−1 is attributed to the D-band of graphene. The intensity ratio of 2D to G bands (I2D/IG) was measured to be around 3 and the 2D band is symmetric, both are characteristics of a monolayer graphene21. The HR-TEM image shows a graphitic crystalline structure (Figure 1c). The corresponding selected area electron diffraction (SAED) pattern exhibits a hexagonal pattern and the intensity of {1-100} is stronger than that of {1-210} (inset of Figure 1c), also reflecting the features of monolayer graphene22,23. The scanning electron microscope (SEM) image of graphene on a Cu substrate is demonstrated in Figure S2b. This image shows the surface steps of Cu substrate and the blurry wrinkles of graphene. After transferring to a SiO2/Si substrate, the graphene sheet shows typical wrinkles of single-layer graphene sheet24. The uniform gray region in this image is monolayer graphene sheet and the small dark regions are islands of bilayer or multilayers of graphene24,25. According to Figure S2c, the graphene sheet was estimated to have over 95% area of monolayer structure. The thickness of the graphene film was measured by atomic force microscopy (AFM) to be about 0.79 nm (Figure S2d) and this value is in consistent with that of a monolayer graphene26. The conductivity of the single-layer graphene sheet was measured to be as high as 3 × 106 S m−1, making it suitable for electrochemical studies21.
Discussion
The electrochemistry at either the edge- or plane-electrode was studied by cyclic voltammetry in an aqueous solution of 0.1 M phosphate buffered saline (PBS) containing 0.1 M KCl. This electrolyte was selected to eliminate the pH effect and obtain data that relevant to bio-systems. In the pure electrolyte, the cyclic voltammograms (CVs) for both electrodes are rectangular in shape, indicating a pure capacitive behavior (Figure 2). These CVs were performed in a narrow potential range to avoid unrequired side reactions. The areal specific capacitances of the edge-electrode was estimated to be about 1.0 × 105 μF cm−2 (the thickness of monolayer graphene was estimated to be 1 nm, see Methods section) and this value is over 4 orders of magnitude higher than that of plane-electrode (4 μF cm−2). The graphene edge was formed by mechanical cutting, leading to the formation of structural defects with dangling bonds. These dangling bonds are unstable upon the exposure to air or solution and they can be possibly terminated by oxygen containing groups or adsorbing other reactive species27. The high specific capacitance of the edge is mainly due to the accumulation of ions from electrolyte at its structural defects to form electrochemical double layers28,29.

Capacitive comparison of graphene edge and basal plane.
(a, b) Cyclic voltammograms of the edge- (a) and plane- (b) electrodes in a 0.1 M PBS solution containing 0.1 M KCl at a scan rate of 50 mV s−1.
The charge transfer processes at the edge- and plane-electrodes were further studied by using potassium ferricyanide, K3Fe(CN)6, as an electrochemical probe. Figure 3a and 3c give the CVs of 5 mM K3Fe(CN)6 in the same electrolyte and at different potential scan rates. The CV wave current density for edge- or plane-electrode increases linearly with the square root of scan rate (Figure 3b and 3d), indicating the electrochemical processes are controlled by the diffusion of electroactive species. For a linear diffusion-based system, the wave current (ip) can be given by Randles-Sevcik equation10:

where n is the number of electrons involved in the redox reaction (here, n = 1), A is the area of the electrode, C is the concentration of the electrochemical probe, D is the diffusion coefficient and v is the scan rate. For the plane-electrode, the wave current density (j = ip/A) was measured to be 2.2 × 10−4 A cm−2 at a scan rate of 5 mV s−1. This current density is in good agreement with both the theoretically calculated value (j = 2.6 × 10−4 A cm−2) and the wave current density acquired at a conventional glassy carbon (GC) electrode (2.2 × 10−4 A cm−2, Figure S3). These results confirm that the electrochemical reaction on plane-electrode is controlled by linear diffusion and the electrochemical property of graphene basal plane is similar to that of conventional GC. For the edge-electrode, the current density calculated from equation 1 is 2.6 × 10−4 A cm−2 and this value is about 400 times lower than the experimental CV wave current density (0.11 A cm−2). The edge-electrode is an ultramicroband with a width around 1 nm and a length in millimeter scale; thus the diffusion process at its surface is likely different from that at the macroscopic plane-electrode. Actually, the CV curves recorded for the edge-electrode exhibit a quasi-sigmoidal shape even at a high scan rate of 100 mV s−1 (Figure 3a). At a slow scan rate of 5 mV s−1, a convergent diffusion mode is dominating (Inset of Figure 3a)30,31. Convergent diffusion resulted in faster mass transport and stronger wave current density32. The steady current (is) for an ultramicroband electrode can be approximately calculated by equation 230:

where, L and w are the length and width of edge-electrode, respectively. The other terms have their usual electrochemical meanings (cf. equation 1). Accordingly, the steady current density (j = is/Lw) was calculated to be 10 A cm−2 at a scan rate of 5 mV s−1, which is about 2 orders of magnitude higher than the experimental value (j = 0.11 A cm−2). A possible explanation for the theoretical overestimation given by equation 2 is the limitation of its diffusion law. For example, the flux in memory diffusion that based on the Fick's first law is generated with a delay from the formation of concentration gradient33,34. Furthermore, a fast electrochemical process can cause a delay of diffusion; the actual diffusion velocity is much slower than the theoretical value. As a result, the concentration of electrochemical probe at the surface of edge-electrode is much lower than that derived by using the theoretical mode of convergent diffusion. It was reported that such deviation was strongly dependent on the size of electrode. For an electrode with a diameter larger than 10 μm, this effect is negligible. For an electrode with a diameter as small as 0.11 μm, however, the experimental current density was more than 1 order of magnitude lower than the theoretical value33. In our case, the edge-electrode is only one-atom thick, allowing only one ion to access each site of its surface in the through-thickness direction. The quasi-sigmoidal shaped CV curves (Figure 3a) together with the linear diffusion behavior (Figure 3b) reflect that the redox species diffuse to the surface of the edge-electrode from different directions in a semispherical profile, while only one direction is effective for electrochemical reaction. Nevertheless, the theoretical overestimation caused by a surface blockage effect and/or the adsorption of side products cannot be extensively ruled out30,35.

Electrochemistry of ferricyanide at the graphene edge- or plane-electrode.
(a, b) CVs of 5 mM K3Fe(CN)6 at the graphene edge-electrode at different scan rates (a) and the plot of saturated current density versus square root of scan rate (b); inset of (a) is the CV recorded at 5 mV s−1. (c, d) CVs of 5 mM K3Fe(CN)6 at the graphene plane-electrode at different scan rates (c) and the plot of saturated current density versus square root of scan rate (d); inset of (b) is the CV recorded at 5 mV s−1. The electrolyte = 0.1 M PBS + 0.1 M KCl.
In spite of the theoretical overestimation discussed above, the observed steady wave current density of ferricyanide at the edge-electrode is at least 2 orders of magnitude stronger than that measured at the plane-electrode (Figure 3), indicating a much faster electron transfer. Indeed, the heterogenous electron transfer rate can be accelerated by increasing the density of electronic states (DOS), relating to the amount of edge defects36. The convergent diffusion also induced a faster mass transfer to contribute a stronger wave current density10. Our findings have also been supported by a recent work on the electrochemistry at the nanopore made of single-layer CVD-graphene sandwiched between Al2O3 dielectric layers10. In that case, the nanopores were fabricated by electron beam sculpting and the pore diameters were in the range of 5 to 20 nm. The electrode lengths equal to the pore circumferences; thus, they are in nanometer scale and significantly smaller than that of the edge-electrode used in this study (1–2 mm). As a result, the influence of convergent diffusion is more prominent, leading to a much stronger current density (on the order of 104 A cm−2). This high current density is also partly attributed to the high concentration of electrochemical probe (1 M KCl). In comparison with this previous study, our method is more conventional and simpler, avoiding the expensive and complicated lithography process.
The electrocatalytic activities of the edge- and plane-electrodes have been studied by using 3 electrochemical probes, including ascorbic acid (AA), beta-nicotinamide adenine dinucleotide (NADH) and oxygen. AA plays an important role in health care for human beings,especially essential to skin, connective tissues and immune systems37. NADH is an important biomolecule that has wide applications in dehydrogenase-based biosensors15. Figure 4a, b show the typical CV curves of 5 mM AA in 0.1 M PBS + 0.1 M NaCl electrolyte. At the edge-electrode, the CV exhibits an oxidation wave centered at ca. 0.30 V (Figure 4a), while the corresponding wave peak at the plane-electrode is positively shifted to 0.41 V (Figure 4b). The CV wave peak of NADH at the edge-electrode is located at ca. 0.44 V (Figure 4c). In comparison, the corresponding wave peak at the plane-electrode is also positively shifted to ca. 0.64 V (Figure 4d). It should be noted here that the peak current on the second CV cycle of NADH oxidation was observed to be much weaker than that on the first cycle (56% lower for the edge and 15% lower for the basal plane, Figure S4). This is possibly due to the adsorption of NAD+, the oxidized product of NADH, on the electrode surfaces38. The electrochemical results described above indicate that the graphene edge is much superior to its basal plane on the electrocatalytic oxidation of AA and NADH. It has been reported that the oxidation of AA was an inner sphere reaction and the electron transfer kinetics was highly sensitive to the surface properties of electrode20. The surface of plane-electrode has a nearly perfect graphitic structure. The more defective sites and oxygen containing groups on the surface of edge-electrode greatly reduced the over-potential for AA oxidation. Furthermore, NAD+ ions are favor to adsorb on defects, especially those with carboxyl groups38. The much stronger passivation of the edge-electrode caused by the adsorption of NAD+ ions further confirmed that the edge defects are responsible for the electrocatalytic activity of graphene.

Electrochemistry of AA, NADH or O2 at graphene edge- or plane-electrode.
(a-f) CVs of (a, b) 5 mM AA, (c, d) 5 mM NADH or (e, f) O2 at graphene edge-electrode (a, c, e); or plane-electrode (b, d, f). Electrolyte for (a–d) = 0.1 M PBS + 0.1 M KCl and that for (e, f) = 0.1 M KOH; scan rate = 50 mV s−1.
In addition to the oxidation reactions of two biomolecules, AA and NADH, the oxygen reduction reaction (ORR) has also been studied at both the graphene edge- and plane-electrodes. In this case, the CVs were performed in 0.1 M KOH solution at a scan rate of 50 mV s−1. Both electrodes showed featureless CV curves in N2 saturated solutions within the potential range of −0.9 to 0 V (Figure 4e and 4f). After saturating the solutions with O2, the CVs at both electrodes exhibited strong waves of ORR. The CVs of edge- and plane-electrodes did not have any obvious difference in their onset potentials (−0.22 V vs SCE), while a large difference in their peak potentials was observed. The CV of edge-electrode has an ORR peak centered at −0.48 V (Figure 4e), whereas that at the plane-electrode is negatively shifted to −0.71 V (Figure 4f). Density function theory (DFT) calculations have shown that dangling bonds at the edge of a graphene sheet have strong ability for the adsorption of O239. The dissolved O2 molecules were selectively adsorbed on defects and changed the graphene edge to a p-type semiconductor. As a result, the polarity of the edge is much higher than that of the basal plane. The high polarity along with the existence of oxygen containing groups at graphene edge make the edge-electrode more hydrophilic in alkaline condition40,41, providing a stronger affinity to the electrolyte and dissolved oxygen. The performances of both electrodes on ORR also confirm that the edge of graphene possesses a much stronger electrocatalytic activity than that of graphene basal plane.
In summary, electrodes based on graphene edge and basal plane have been fabricated by region-specific coating CVD-grown monolayer graphene sheet with epoxy resin. Electrochemical studies indicate that the specific capacitance of graphene edge is 4 orders of magnitude higher than that of graphene basal plane. Furthermore, the CV waves of the graphene edge-electrode are quasi-sigmoidal in shape, following a convergent diffusion mode. However, the plot of steady current density versus square root of scan rate is linear, following a linear diffusion mode. The current density of the CV wave recorded at the edge-electrode is at least 2 orders of magnitude stronger than that recorded at the plane-electrode at a given scan rate because of a higher electron-transfer rate and the convergent diffusion at the ultramicroband edge-electrode. Graphene edge also has a much stronger electrocatalytic activity than graphene basal plane, not only on the oxidation reactions of AA and NADH but also on ORR. These findings demonstrate that the large basal plane of a monolayer graphene sheet is highly conductive while the graphene edge possesses superb capacitive and electrocatalytic properties.
Methods
Monolayer graphene sheets
The monolayer graphene sheets were grown on 25-μm thick Cu foils by CVD technique (Alfa Aesar, #13382)23,42. The details are described as follows. A Cu foil (cleaned by dilute acetic acid and acetone before use) was put into a tri-zone furnace (Lindberg Blue M HTF55667C) with 1-inch quartz tube. After filling with H2 (20 sccm at 0.30 Torr), the temperature in the furnace was increased from room temperature to 900°C within 25 min and then to 1000°C within another 10 min. After a 20 min-annealing process, a mixture of 10 sccm CH4 and 5 sccm H2 was introduced at 0.35 Torr for the growth of graphene sheet at 1000°C for 30 min. Then the furnace was firstly cooled to 700°C at a rate of 15°C min−1 and finally spontaneously cooled down to room temperature. The gas flow was kept alive throughout the cooling process.
Preparation of the electrodes
3% solution of PMMA with a molar mass of 996 K (Sigma Aldrich, #182265, dissolved in ethyl lactate) was spin-coated on a graphene/Cu sheet at 2000 rpm (runs per minute) for 60 s and dried at 170°C for 2 min. Then, air plasma (Femto, Diener Electronics) was used to remove graphene on the back side of Cu foil. The edge-electrode was fabricated by coating the middle region of a graphene/Cu foil with ER (Nanjing Xilite Adhesive CO., LTD., China), leaving its two ending parts uncovered. Successively, the electrode was heated at 60°C for 1 h to solidify ER. Thereafter one end of this electrode was immersed in a FeCl3 solution (aqueous solution of 0.1 g mL−1 FeCl3 + 0.1 mol L−1 HCl) to regionally etch away the Cu substrate and expose graphene sheet and the other end was used for connecting potentiostat for subsequent electrochemical measurements. After rinsed with distilled water for several times, the graphene end without Cu substrate was further coated with ER and cut vertically to expose its cross-section. The both corner regions of the exposed graphene edge were further coated with ER to leave only a narrow middle region (1.0–2.0 mm) of the graphene edge. The plane-electrode was prepared by partly coating the graphene/Cu sheet with ER, leaving one end free from the ER coating for electrical connection with potentiostat and an ER-free window in its middle region. The Cu in the window was etched in FeCl3 solution and rinsed with distilled water several times. Finally, the window wall was further coated with ER to avoid the percolation of electrolyte (Figure S1).
Surface areas of electrodes
The surface area of an electrode was calculated by the multiplication of its length and width. The length and width of a plane-electrode were measured by a vernier caliper or reading from its SEM image. The length of an edge-electrode was also accurately measured by these techniques. Its width is theoretically identical to the thickness of a single-layer graphene sheet (0.79 nm). Taking the wrinkle structure of graphene edge and its oxygenated groups into account, we evaluated the width of an edge-electrode to be around 1 nm.
Characterizations
All electrochemical tests were performed on an electrochemical analyzer (CHI 660D potentiostat-galvanostat, CH Instruments Inc.) at room temperature. The graphene electrode, a Pt foil and a SCE electrode were used as the working, counter and reference electrodes, respectively. These 3 electrodes were parallel positioned in the elecrochemical cell (Figure S5). Raman spectra were recorded on a Renishaw Raman microscope (RM2000) with a 514-nm laser at a power density of 4.7 mW. Scanning electron micrographs (SEM) were performed on a field-emission scanning electron microscope (Sirion-200, Japan). High-resolution transmission electron microscope (HRTEM) images, selected area electron diffraction (SAED) pattern were taken by Tecnai G2 F20S-Twin HRTEM. AFM image was taken out by using a SPM-9600 microscope (Shimadzu).
References
Li, W., Tan, C., Lowe, M. A., Abruna, H. D. & Ralph, D. C. Electrochemistry of individual monolayer graphene sheets. ACS Nano 5, 2264–2270 (2011).
Huang, X., Qi, X. Y., Boey, F. & Zhang, H. Graphene-based composites. Chem. Soc. Rev 41, 666–686 (2012).
Liu, C. G., Yu, Z. N., Neff, D., Zhamu, A. & Jang, B. Z. Graphene-based supercapacitor with an ultrahigh energy density. Nano Lett. 10, 4863–4868 (2010).
Geim, A. K. & Novoselov, K. S. The rise of graphene. Nat. Mater. 6, 183–191 (2007).
Brownson, D. A. C., Gomez-Mingot, M. & Banks, C. E. CVD graphene electrochemistry: biologically relevant molecules. Phys. Chem. Chem. Phys. 13, 20284–20288 (2011).
Zhu, Y. W. et al. Carbon-based supercapacitors produced by activation of graphene. Science 332, 1537–1541 (2011).
Sheng, K., Sun, Y., Li, C., Yuan, W. & Shi, G. Ultrahigh-rate supercapacitors based on eletrochemically reduced graphene oxide for ac line-filtering. Sci. Rep. 2, 247; 10.1038/srep00247 (2012).
Davies, T. J., Hyde, M. E. & Compton, R. G. Nanotrench arrays reveal insight into graphite electrochemistry. Angew. Chem.-Int. Edit. 44, 5121–5126 (2005).
Pumera, M., Ambrosi, A., Bonanni, A., Chng, E. L. K. & Poh, H. L. Graphene for electrochemical sensing and biosensing. TrAC, Trends Anal. Chem. 29, 954–965 (2010).
Banerjee, S. et al. Electrochemistry at the edge of a single graphene layer in a nanopore. ACS Nano 7, 834–843 (2012).
Brownson, D. A. C. & Banks, C. E. CVD graphene electrochemistry: the role of graphitic islands. Phys. Chem. Chem. Phys. 13, 15825–15828 (2011).
Banks, C. E., Davies, T. J., Wildgoose, G. G. & Compton, R. G. Electrocatalysis at graphite and carbon nanotube modified electrodes: edge-plane sites and tube ends are the reactive sites. Chem. Commun. 829–841 (2005).
Anne, A. S., Cambril, E., Chovin, A., Demaille, C. & Goyer, C. D. Electrochemical atomic force microscopy using a tip-attached redox mediator for topographic and functional imaging of nanosystems. ACS Nano 3, 2927–2940 (2009).
Patel, A. N. et al. A New View of Electrochemistry at Highly Oriented Pyrolytic Graphite. J. Am. Chem. Soc. 134, 20117–20130 (2012).
Banks, C. E. & Compton, R. G. Exploring the electrocatalytic sites of carbon nanotubes for NADH detection: an edge plane pyrolytic graphite electrode study. Analyst 130, 1232–1239 (2005).
Banks, C. E. & Compton, R. G. New electrodes for old: from carbon nanotubes to edge plane pyrolytic graphite. Analyst 131, 15–21 (2006).
Chou, A., Bocking, T., Singh, N. K. & Gooding, J. J. Demonstration of the importance of oxygenated species at the ends of carbon nanotubes for their favourable electrochemical properties. Chem. Commun. 842–844 (2005).
Miller, T. S., Ebejer, N., Gueell, A. G., Macpherson, J. V. & Unwin, P. R. Electrochemistry at carbon nanotube forests: sidewalls and closed ends allow fast electron transfer. Chem. Commun. 48, 7435–7437 (2012).
Gooding, J. J. et al. Protein electrochemistry using aligned carbon nanotube arrays. J. Am. Chem. Soc. 125, 9006–9007 (2003).
Gong, K., Chakrabarti, S. & Dai, L. M. Electrochemistry at carbon nanotube electrodes: Is the nanotube tip more active than the sidewall? Angew. Chem.-Int. Edit. 47, 5446–5450 (2008).
Suk, J. W. et al. Transfer of CVD-grown monolayer graphene onto arbitrary substrates. ACS Nano 5, 6916–6924 (2011).
Levendorf, M. P., Ruiz-Vargas, C. S., Garg, S. & Park, J. Transfer-free batch fabrication of single layer graphene transistors. Nano Lett. 9, 4479–4483 (2009).
Yan, K., Peng, H., Zhou, Y., Li, H. & Liu, Z. Formation of bilayer bernal graphene: layer-by-layer epitaxy via chemical vapor deposition. Nano Lett. 11, 1106–1110 (2011).
Kholmanov, I. N. et al. Improved electrical conductivity of graphene films integrated with metal nanowires. Nano Lett. 12, 5679–5683 (2012).
Kalita, G., Wakita, K. & Umeno, M. Low temperature growth of graphene film by microwave assisted surface wave plasma CVD for transparent electrode application. RSC Adv. 2, 2815–2820 (2012).
Wang, H. et al. Synthesis of Boron-Doped Graphene Monolayers Using the Sole Solid Feedstock by Chemical Vapor Deposition. Small 9, 1316–1320 (2013).
Wang, X., Tabakman, S. M. & Dai, H. Atomic Layer Deposition of Metal Oxides on Pristine and Functionalized Graphene. J. Am. Chem. Soc. 130, 8152–8153 (2008).
Randin, J. P. & Yeager, E. Differential capacitance study of stress/annealed pyrolytic graphene electrodes. J. Electrochem. Soc. 118, 711–714 (1971).
Yoo, J. J. et al. Ultrathin planar graphene supercapacitors. Nano Lett. 11, 1423–1427 (2011).
Wehmeyer, K. R., Deakin, M. R. & Wightman, R. M. Electroanalytical properties of band electrodes of submicrometer width. Anal. Chem. 57, 1913–1916 (1985).
Metters, J. P., Kadara, R. O. & Banks, C. E. Electroanalytical properties of screen printed graphite microband electrodes. Sensor. Actuat. B-Chem. 169, 136–143 (2012).
Le Drogoff, B., El Khakani, M. A., Silva, P. R. M., Chaker, M. & Vijh, A. K. Effect of the Microelectrode geometry on the diffusion behavior and the electroanalytical performance of Hg-electroplated iridium microelectrode arrays intended for the detection of heavy metal traces. Electroanalysis 13, 1491–1496 (2001).
Aoki, K., Ouyang, C., Zhang, C., Chen, J. & Nishiumi, T. Comparison of diameters of disk microelectrodes obtained from microscopes with those evaluated from steady-state currents. Int. J. Electrochem. Sc. 7, 5880–5892 (2012).
Aoki, K. & Xiang, C. Relaxation Time of Memorial Diffusion by Chronoamperometry at a Twin-electrode. J. Phys. Chem. C 111, 15433–15439 (2007).
Chen, S. L. & Kucernak, A. Fabrication of carbon microelectrodes with an effective radius of 1 nm. Electrochem. Commun. 4, 80–85 (2002).
Brownson, D. A. C., Kampouris, D. K. & Banks, C. E. Graphene electrochemistry: fundamental concepts through to prominent applications. Chem. Soc. Rev 41, 6944–6976 (2012).
Han, D. F., Shan, C. S., Guo, L. P., Niu, L. & Han, D. X. Electro-oxidation of ascorbic acid on PVP-stabilized graphene electrode. Chem. Res. Chin. Univ. 26, 287–290 (2010).
Pumera, M. et al. A mechanism of adsorption of beta-nicotinamide adenine dinucleotide on graphene sheets: experiment and theory. Chem.-Eur. J. 15, 10851–10856 (2009).
Acik, M. & Chabal, Y. J. Nature of graphene edges: a review. Jpn. J. Appl. Phys. 50, 070101–070116 (2012).
Jeon, I.-Y. et al. Large-scale production of edge-selectively functionalized graphene nanoplatelets via ball milling and their use as metal-free electrocatalysts for oxygen reduction reaction. J. Am. Chem. Soc. 135, 1386–1393 (2012).
Wang, X. L. et al. Solution-processable graphene nanomeshes with controlled pore structures. Sci. Rep. 3, 1996; 10.1038/srep01996 (2013).
Li, X. S. et al. Large-area synthesis of high-quality and uniform graphene films on copper foils. Science 324, 1312–1314 (2009).
Acknowledgements
This work was supported by national basic research program of China (973 Program, 2012CB933402, 2013CB933000), natural science foundation of china (51161120361, 91027028) and NSFC-NSF (DMR-1106160).
Author information
Authors and Affiliations
Contributions
G.S. and L.D. developed the concept, designed experiments and co-wrote the paper. W.Y. performed experiments, analysed data and wrote the paper. Y.Z., H.P. and Z.L. prepared the CVD graphene sheets. Y.L., C.L., H.P., J.Z. and Z.L. gave helps in the experiments and co-wrote the paper. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Yuan, W., Zhou, Y., Li, Y. et al. The edge- and basal-plane-specific electrochemistry of a single-layer graphene sheet. Sci Rep 3, 2248 (2013). https://doi.org/10.1038/srep02248
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02248
This article is cited by
-
Low-temperature synthesis of colloidal few-layer WTe2 nanostructures for electrochemical hydrogen evolution
Discover Nano (2023)
-
Relationship between pore structure of N-doped 3D porous graphene and electrocatalytic performance of oxygen reduction in zinc-air battery
Journal of Central South University (2023)
-
Electrochemical characterization and degradation of carbon fibre reinforced polymer in quiescent near neutral chloride media
npj Materials Degradation (2022)
-
Edge engineering in chemically active two-dimensional materials
Nano Research (2022)
-
Glucose measurement via Raman spectroscopy of graphene: Principles and operation
Nano Research (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
