
Abstract
Wernicke's encephalopathy (WE) is characterized by lesions in thalamus, hypothalamus (including mammillary nuclei), and inferior colliculi, results in serious disabilities, has an etiology of thiamine deficiency, is treatable with thiamine, and occurs most commonly with alcoholism. Despite decades of study, whether alcohol exposure exacerbates the neuropathology or retards its resolution remains controversial. To examine patterns of brain damage and recovery resulting from thiamine deprivation with and without alcohol exposure, we conducted in vivo magnetic resonance imaging (MRI) and magnetic resonance spectroscopy (MRS) at 3 T in alcohol-preferring (P) rats, which had voluntarily consumed large amounts of alcohol before thiamine manipulation. A total of 18 adult male P rats (nine alcohol-exposed) received a thiamine-deficient diet for 2 weeks: 10 (five alcohol-exposed) received intraperitoneal (i.p.) pyrithiamine (PT) and eight (four alcohol-exposed) received i.p. thiamine supplementation. Neurological signs developed by day 14. Rats were scanned before thiamine depletion and 18 and 35 days after thiamine repletion. Two-dimensional J-resolved MRS single-voxel spectra with water reference were collected in a voxel subtending the thalamus; metabolite quantification was corrected for voxel tissue content. MRI identified significant enlargement of dorsal ventricles and increase in signal intensities in thalamus, inferior colliculi, and mammillary nuclei of PT compared with thiamine-treated (TT) groups from MRI 1–2, followed by significant normalization from MRI 2–3 in thalamus and colliculi, but not mammillary nuclei and lateral ventricles. Voxel-by-voxel analysis revealed additional hyperintense signal clusters in the dorsal and ventral hippocampus and enlargement of the fourth ventricle. MRS showed a significant decline and then partial recovery in thalamic N-acetylaspartate, a marker of neuronal integrity, in PT compared with TT rats, with no change detected in creatine, choline, or glutamate. PT rats with prior alcohol exposure exhibited attenuated recovery in the thalamus and arrested growth of the corpus callosum; further, two of the five alcohol-exposed PT rats died prematurely. Parenchymal and ventricular changes with thiamine manipulation concur with human radiological signs of WE. The enduring macrostructural and neurochemical abnormalities involving critical nodes of Papez circuit carry liabilities for development of amnesia and incomplete recovery from other cognitive and motor functions subserved by the affected neural systems.
Similar content being viewed by others
INTRODUCTION
More than a century ago, Wernicke described features of an encephalopathy that selectively affects oculomotor function, gait, and mentation as it relates to orientation to time and place and is accompanied by hemorrhagic pathology in periventricular gray matter structures (as noted in Victor et al, 1971, 1989). The rigorous neurological and neuropathological analysis of cases exhibiting symptoms of Wernicke's encephalopathy (WE) by Victor, Adams, and Collins (Victor et al, 1971, 1989) identified structures with the highest incidence of CNS pathology – the thalamus and mammillary nuclei of the hypothalamus, periaqueductal gray matter, brainstem nuclei, particularly the superior and inferior colliculi, and anterior cerebellar vermis. Recognizing that WE commonly accompanied poor nutritional status of hyperemesis gravidarum, starvation, anorexia nervosa, or alcoholism, early experimentation and astute observation pinpointed thiamine depletion as the cause of WE (Bender and Schilder, 1933; Jolliffe et al, 1941). Alcoholics are at special risk for thiamine deficiency because of the poor diet associated with their lifestyle and because chronic alcoholism compromises thiamine absorption from the gastrointestinal tract, impairs storage, and may reduce thiamine phosphorylation, essential for cellular function (Lieber, 2003a; Martin et al, 2003; Thomson et al, 1987; Todd and Butterworth, 1999). Parenteral thiamine treatment given in the early symptomatic stages largely reverses the classical neurological signs of ophthalmoplegia, nystagmus, ataxia of gait, and amnesia for new information (Caine et al, 1997; Victor et al, 1971, 1989), although severe or undertreated cases may show enduring memory impairment and ataxia defining Korsakoff's syndrome (Butters and Cermak, 1980; Kopelman, 1995; Talland, 1965; Victor et al, 1959). Neuroradiological studies of WE since the 1970s report in vivo signs of hyperintensity in thalamus, periventricular gray matter, inferior colliculi, and occasionally in cerebellum, observations consistent with post-mortem reports (eg, Baker et al, 1999; Harper and Kril, 1988, 1990; Kril et al, 1997; Torvik et al, 1982, 1986), with partial lesion resolution with treatment. Given the wide variety of precipitating conditions and the devastating morbidities and premature mortality associated with this treatable condition (cf, Harper and Butterworth, 1997; Harper et al, 1995, 1998), it remains clinically relevant to have the means to identify the neuroradiological signature of thiamine deficiency in an animal model of WE with a known etiology and to track the brain lesion for neuronal damage and treatment-associated recovery, as is possible with quantitative in vivo magnetic resonance spectroscopy (MRS).
The effects of thiamine deficiency on brain morphology have been well documented in patients with chronic alcoholism, which itself produces widespread shrinkage of brain tissue (Cardenas et al, 2005; Fein et al, 2002; Jernigan et al, 1991; Pfefferbaum et al, 1992) and attendant deficits in motor and cognitive function that can persist even after months of abstinence along with adequate nutrition (eg, Eckardt et al, 1998; Fein et al, 1990; Parsons, 1993; Sullivan et al, 2000c). Although alcohol-related brain abnormalities are partially reversible with prolonged sobriety (Carlen et al, 1978; Gazdzinski et al, 2005; Mann et al, 1999; O'Neill et al, 2001; Parks et al, 2002; Pfefferbaum et al, 1995, 1998; Schroth et al, 1988), cortical gray and white matter may sustain long-term volume shrinkage and even loss (Jernigan et al, 1991; Pfefferbaum et al, 1992), especially in the prefrontal cortex (De Bellis et al, 2005) of older alcoholics (Cardenas et al, 2005; Pfefferbaum et al, 1997). Like amnesic patients with Wernicke–Korsakoff syndrome, non-amnesic alcoholics also have notable volume shrinkage of the mammillary bodies (Davila et al, 1994; Shear et al, 1996; Sullivan et al, 1999; but see Charness and DeLaPaz, 1987), anterior hippocampus (Agartz et al, 1999; Sullivan and Marsh, 2003; Sullivan et al, 1995), thalamus (Sullivan et al, 2003), and cerebellum (Sullivan et al, 2000a, 2000b), but the volume shrinkage is substantially less than in alcoholics with WE or KS (cf, Blansjaar et al, 1992; Charness, 1993, 1999; Mulholland et al, 2005; Sullivan, 2000). Because of the edematous nature of WE lesions, magnetic resonance imaging (MRI) methods sensitive to fluid changes in tissue have revealed bilaterally distributed hyperintensities in medial thalamus, mammillary bodies, and periaqueductal gray matter (eg, non-alcoholics: Chu et al, 2002; Doraiswamy et al, 1994; Unlu et al, 2006; Zhong et al, 2005) (alcoholics: Schroth et al, 1991).
Residual neural integrity in the area of damaged tissue can be assessed with in vivo proton MRS. In alcoholism uncomplicated by WE, MRS provides evidence for abnormally low peaks of N-acetylaspartate (NAA), a marker for mature viable neurons, or choline (Cho), an index of membrane turnover, in frontal, parietal, or cerebellar regions within a month of withdrawal (Durazzo et al, 2004; Fein et al, 1994; Jagannathan et al, 1996; Schweinsburg et al, 2003, 2001; Seitz et al, 1999) followed by improvement in NAA or Cho, suggesting neuronal recovery (Bendszus et al, 2001; Ende et al, 2005; Martin et al, 1995; Parks et al, 2002). In WE, whether preceded by alcoholism or other precipitating condition, an early deficit in NAA can improve, more so in the thalamus than cerebellum (Murata et al, 2001), a recovery pattern noted earlier by Victor et al (1971, 1989). A number of these MRS studies, however, used metabolite ratios rather than absolute concentrations, typically relative to creatine (Cr), to quantify target metabolites, assuming that Cr is robust to CNS aging or insult and therefore remains stable. Indeed, several MRS studies report lack of significant deficits in uncomplicated alcoholics when regional measures of NAA were expressed as a function of the amount of underlying tissue (Mason et al, 2006; Meyerhoff et al, 2004; O'Neill et al, 2001; Pfefferbaum et al, 2005; Schweinsburg et al, 2000). In the Pfefferbaum et al study (2005), where NAA was expressed as a ratio of Cr, an apparent NAA deficit was present, but, it was as much attributable to a lower NAA peak as it was to a higher Cr peak in the alcoholics than controls. Studies of aging and dementia that calculated absolute metabolite values have documented that Cr can change with age (by about 15% higher value in 70-year-olds compared with 25-year-olds observed in Pfefferbaum et al, 1999b) and disease (with similar gray matter elevation in Alzheimer's disease observed in Pfefferbaum et al, 1999a), thus challenging the constancy assumption regarding Cr. An additional issue is the need to express metabolite values as a function of the amount of tissue in the acquired voxel. Again, MRS studies of normal aging have shown that NAA does not decline in normal aging when expressed as a function of the amount of the underlying gray matter, which does lose volume with aging (Adalsteinsson et al, 2000; Pfefferbaum et al, 1999b).
Whether nutritional deficiency, including that of thiamine, is a critical mechanism in any or all of alcoholism-related brain volume and neurochemical abnormalities remains a question in human studies, given the complex, natural course of alcoholism and the necessity of relying on personal history for recounting nutritional status during bouts of uncontrolled, excessive alcohol drinking. These issues can be addressed systematically with longitudinal MRI and MRS studies using animal models of alcoholism and nutritional manipulation.
In rodent studies of thiamine deficiency, neurodegenerative evidence has been observed in the frontal cortex and corpus callosum (Savage et al, 2000), cerebellum (eg, Dlugos and Pentney, 1997; Pentney and Dlugos, 2000; Pentney and Quackenbush, 1990; Rintala et al, 1997), and locus coeruleus (Lu et al, 1997). Initial MRI studies, conducted at 1.5 T, used thiamine deficiency models of WE (Pentney et al, 1993) and reported increased volume of lateral ventricles followed by normalization with a thiamine-enriched diet (Acara et al, 1995). Such neuropathology is accelerated with the dietary thiamine antagonist, pyrithiamine (Langlais and Savage, 1995; Langlais and Zhang, 1997; Lee et al, 2001, 1995). Glucose administration to rats made thiamine deficient with pyrithiamine treatment produced impairment of the blood–brain barrier, observed on T1-weighted images (used for tissue depiction) collected at high scanner field strength (4.7 T) (Zelaya et al, 1995), and hyperintensities in the hippocampus as well as in the thalamus, hypothalamus, collicular bodies, observed on T2-weighted images (used for fluid depiction), that endured for at least 1 month (Jordan et al, 1998). These structural changes were accompanied by a significant increase in the resonance reflecting the combination of glutamate, glutamine, and GABA detected with MRS (Rose et al, 1993). Another MRS study of thiamine deficiency in the rat revealed an early decline and a post-thiamine repletion increase in Cho/NAA, with the interpretation that the Cho increased whereas NAA remained unchanged (Lee et al, 1995). A longitudinal MRS study (performed at 4.7 T) of rats chronically exposed to alcohol for 60 weeks observed a rise in the Cho/NAA ratio that peaked at week 16 of exposure followed by a decline to abnormally low levels after week 44 (Lee et al, 2003).
Using quantitative MRI acquired at 3 and 4.7 T, we observed significant growth in the corpus callosum, cerebellum, and hippocampus of alcohol-naïve adult selectively bred alcohol-preferring (P) rats (Li et al, 1979) over 1-year period during adulthood, from age 88 to 452 days old (Sullivan et al, 2006). Additionally, alcohol consumption by their cohorts, which were amply vitaminized throughout alcohol exposure, attenuated brain growth, particularly of the corpus callosum (Pfefferbaum et al, 2006). The present study examined the short-term and long-term effects of thiamine depletion in the 18 rats used in these growth and alcohol exposure studies (Sullivan et al, 2006). The experimental design involved four groups, all on a thiamine-deficient diet: two with pyrithiamine treatment (one group with and one group without prior alcohol exposure); and two with thiamine replacement (one group with and one group without prior alcohol exposure) (Table 1). Using structural MRI and proton MRS with metabolite concentrations corrected for tissue fraction, we tested the hypotheses that (1) in the early post-treatment period, the thiamine-deficient groups would exhibit the signature lesions of WE, involving the thalamus, inferior colliculi, and mammillary body nuclei and dilatation of the lateral and fourth ventricles; (2) in the later post-treatment with thiamine replacement, the structural lesions would show partial resolution on MRI but continued evidence for cell damage on MRS; (3) lesion resolution would be less complete in thiamine-deficient animals with prior chronic alcohol exposure; and (4) thiamine deprivation would exacerbate corpus callosum thinning in alcohol-exposed rats (cf, Langlais and Zhang, 1997).
MATERIALS AND METHODS
Animals
All rats were obtained from the breeding colony at the Indiana University Medical Center (Indianapolis, IN). The P, from an outbred Wistar foundational stock, rat has undergone over 50 generations of selection for a preference of 10% (v/v) alcohol over concurrently available water. As young adults (approximately 70 days old), the rats were shipped by air to SRI International (Menlo Park, CA), where they remained thereafter and were available for brain imaging. Rats were single housed in plastic tubs with water and food (vitamin- and mineral-enriched Teklad mouse and rat diet #7001; Madison, WI) freely available in the home tubs at all times. The four study groups comprised a cohort of 18 adult male P rats, nine of which had exposure to alcohol under four exposure schedules over the year before the present study; the remaining nine rats were controls, which had never received alcohol. The nine rats never exposed to alcohol had been part of longitudinal MRI studies of normal brain growth (Sullivan et al, 2006) and all 18 rats (nine controls and nine alcohol-exposed) were subjects in a longitudinal study examining the effects of chronic alcohol consumption on brain structure (Pfefferbaum et al, 2006). Selection for group assignment, in the present study, took into account alcohol exposure, litter of origin, body weight, and brain volume, which was determined from the MRI taken before starting thiamine deficiency, that is, the MRI that served as the baseline for the current experiment. At the outset of this experiment, the rats were 452 days old, and the four groups did not differ significantly in body weight (F(3,17)=2.573, n.s.) or brain volume (F(3,17)=0.331, n.s.). Further, the two groups with prior alcohol exposure did not differ in the total amount of alcohol consumed across their 1 year of access (mean±SD, for the pyrithiamine/alcohol group=292.2±16.19 g/kg and for the thiamine/alcohol group=308.8±18.0 g/kg; t(7)=1.45, n.s.).
Thiamine Deprivation Treatment
Following the protocol employed in prior studies (Ciccia and Langlais, 2000; Langlais and Zhang, 1997; Pitkin and Savage, 2001), all rats received thiamine-deficient chow (Harlan-Teklad, TD81029). Ten rats (five alcohol-exposed and five non-exposed) received daily IP injections of pyrithiamine hydrobromide (Aldrich Chemicals) administration (0.25 mg/kg). The remaining four rats from each of the control and alcohol-exposed groups received daily IP injections of thiamine (0.40 mg/kg). The treatment protocol was carried out for 14 consecutive days (pyrithiamine was ended 1 day earlier in one rat with a 90-g weight loss and suspicion of blindness). At the end of the thiamine deprivation period, regular thiamine-enriched chow was resumed for all animals (vitamin- and mineral-enriched Teklad mouse and rat diet #7001; Madison, WI), and 2 weeks later the 10 pyrithiamine-treated animals were administered 100 mg/kg thiamine intraperitoneally (i.p.).
Scanning Schedule and General Scanning Procedures
MRI and MRS protocols were conducted three times, each taking 90–120 min. The baseline sessions occurred 60 days for MRI and 44 days for MRS before the initiation of thiamine deprivation; post-treatment MRI and MRS sessions were conducted on the same day, 18 days and 35 days after re-initiation of thiamine-fortified diet. Two rats in the pyrithiamine/alcohol group died (age 556 and 564 days) before the second post-treatment scans.
MR Scanning Procedures and Processing
MRI and MRS data were acquired on a clinical 3 T GE Signa human MRI scanner fitted with custom RF coils for rats (gradient strength=40 mT/m; slew rate=150 T/m/s; software version VH3) and using standard product sequences. Details of the MRI (Pfefferbaum et al, 2006, 2004a; Sullivan et al, 2006) and MRS procedures (Adalsteinsson et al, 2004, 2006) were described previously and are summarized here. All animal preparation, monitoring, and recovery were conducted according to procedures approved by the Administrative Panel on Laboratory Animal Care and performed by two highly experienced veterinary technicians (RVT, LATg). The rats were anesthetized using 1–3% isoflurane; heart rate, pulse rate, oxygen saturation, and core temperature were monitored continually. Rats were scanned in a custom animal positioning system (John Houseman, Thurso, Caithness, Scotland) for head placement and immobilization.
MRI acquisition
Data were acquired with an eight-rung high-pass birdcage transmit/receive coil. A 21-slice per plane, three-plane localizer (TE=3 ms, TR, FOV=80 mm, 256 × 128 pixel resolution, 0.5 mm slice thickness) was used to define the rostral and caudal extremes of each rat brain; the sagittal view was used for graphical prescription. A dual-echo spin-echo acquisition was graphically prescribed to cover the entire rat brain (3.3 cm) in 66 coronal slices (TE1=16 ms, TE2=50 ms, TR=5 s, FOV=60 mm, 256 × 256 pixel resolution, 0.5 mm slice thickness, 22 min acquisition time) and run three times for improved signal-to-noise ratio. The resulting native spatial resolution was 234 × 234 × 500 μm.
MRI analysis
Brain images were transferred to a workstation where they were analyzed using in-house software routines, written in IDL (Interactive Data Language, Boulder, CO). The images were interpolated in-plane from the acquired 256 × 256 resolution, which corresponds to 234 μm pixel size, to 78 μm pixel size. The center of the brain at the center slice was identified, and interactively, a midsagittal line was drawn. The data were then rotated such that the midsagittal orientation was vertical. The middle 256 × 256 pixels of this centered, rotated volume were extracted, as they were sufficient to contain all brain voxels.
Before averaging the separate acquisitions for each animal within a session, the data were spatially aligned on a slice-by-slice basis to maximize a cross-correlation metric, with no slice shifts larger than 1.2 mm. This procedure reduced blurring and increased conspicuity of tissue margins compared with simple averaging without alignment, thereby reducing any adverse effects of motion during the scan. The two acquired echoes were combined in a weighted average. The brain was then extracted based on nonparametric thresholding and two-dimensional contour filling. Brains were also resampled through-plane into 78 μm isotropic voxels, and a well-aligned, never-treated animal from a pretreatment session was chosen as the template data set to which each individual animal for each session was aligned using a six-parameter rigid-body function (AIR 5.0), thus preserving brain size.
To investigate parenchymal effects of thiamine deficiency, grand average brain images were constructed for intensity-normalized early- and late-echo data for each group separately for pre- and post-treatment conditions, followed by creation of subtraction images, reflecting post-treatment minus pretreatment. From visual inspection of three-dimensional projections (sagittal, coronal, and axial views) of the difference images, three brain regions had obvious signal intensity increases from baseline to first post-treatment scans. Projection of these regions onto pretreatment structural images identified them as the thalamus, mammillary bodies, and inferior colliculi bilaterally (Figure 1). To quantify treatment effects, regions of interest were defined on the grand average with a histogram threshold operator and manual editing. Signal intensities in these three regions from early-echo and late-echo images at all three sessions were quantified, as were T2 relaxation time estimates derived from the dual-echo data.

(a) Slice of a sagittal grand average post–pre difference image showing hyperintense areas in the thalamus and mammillary nuclei in pyrithiamine-treated rats. (b) Slice of an early-echo structural image at the same location as image a. (c) Slice of a coronal grand average post–pre difference image showing bilateral hyperintense areas in the inferior colliculi in the same rat. (d) Slice of an early-echo structural image at the same location as image c.
Manual regional size measurements were made on midsagittal corpus callosum area and coronal images of the cerebellum and dorsolateral ventricles for all MRI sessions using anatomical landmarks (Pfefferbaum et al, 2004a; Sullivan et al, 2006).
MRS acquisition
MRI and MRS data were acquired on separate sessions, 10 days apart pretreatment and within 2 days of each other for the two post-treatment sessions. For the MRS acquisition, a three-plane localizer scan (TE/TR=2.1/54 ms, FOV=80 mm, 256 × 128, 5 mm thick, 10 slices/plane) was used for prescription of coronal fast spin-echo (FSE) images (TE1/TE2/TR=18/90/2200 ms, FOV=40 mm, 256 × 256, 1.0 mm thick, 33 slices). An approximately 0.25 cm3 brain voxel (10 × 5 × 5 mm3) was prescribed on the FSE images encompassing the majority of the rostral–caudal and lateral extents of the thalamus.
Single-voxel two-dimensional (2D) J-resolved MRS spectra were acquired, using 4.7 min acquisitions (Dreher and Leibfritz, 1995; Hurd et al, 1998) (TE1/TR=35/2000 ms, ΔTE=10 ms, 16 TE steps, eight averages per TE step) repeated four times with water suppression and once without water suppression. Water suppression was performed with CHESS (Haase et al, 1985), with flip angles determined by the default GE PRESS sequence. These water suppression settings were found to pass through an adequate amount of residual water (between 10 : 1 and 100 : 1 relative to NAA) to provide phasing of the metabolite spectra for each TE. Each read-out was sampled with 2048 points at a 5 kHz bandwidth.
Spectral processing
The data with partial water suppression were phase corrected based on residual water as described in Webb et al (1994). A zero- and first-order phase terms were extracted and the signal phase unwrapped, followed by a smoothing spline fit to any residual nonlinear phase component, which is then removed. The time-domain data were then apodized with a 6 Hz Gaussian filter, zero filled to 4096 points and Fourier transformed. Residual water and water sidebands were removed from the partially suppressed metabolite data through pure water subtraction (Adalsteinsson et al, 2004; Hurd, 2000). A single linear term spanning the entire spectral bandwidth was then removed based on a fit to data in spectral bins near the highest and lowest frequencies (±2.5 kHz). A final frequency correction step was performed to compensate for small temperature variations during the scans by referencing each frame to NAA at 2.02 p.p.m.
For each 2D acquisition, the TE-averaged spectrum (ie, the average of the spectra from TE values 55–195) was used for quantification of metabolites. This corresponds to only using the centerline from a 2D J-resolved spectrum for the analysis. For each of the four spectra with water suppression, the spectra were aligned at 2.02 p.p.m., based on the maximum peak amplitude of NAA, and then averaged. Peak integration through fitting to a Gaussian line shape was used to quantify the spectral peaks in the TE-averaged spectra. To control for shim variation, the width of the Gaussian fit to NAA was used to determine the line width for all metabolites measured (NAA at 2.02, Cho at 3.25, Cr at 3.01, Cr2 at 3.95, Glu at 2.35 p.p.m.).
The 2D J-resolved MRS spectra with no water suppression were used to estimate the CSF and tissue fraction of the press box with a bi-exponential model to fit the water peak across the 16 TEs. To improve numerical stability of the nonlinear fit, the CSF T2 was fixed at a long T2 value (500 ms) and the remaining short T2 component of tissue determined from a minimization search. The intercept at TE=0 for the two functions was used to estimate the CSF and tissue concentrations in arbitrary units. A regression analysis was performed to assess the influence of the amount of tissue excited in the press box and the effects of changes in scanner sensitivity across animals and sessions on the observed metabolite spectra. Pretreatment metabolite data from all animals and all post-treatment data from the thiamine-treated animals were regressed on the tissue concentration for the same animal and session (Figure 2). Each metabolite and the sum of all five metabolites were positively correlated with the tissue concentration (NAA r=0.905, Cho r=0.840, Cr r=0.809, Cr2 r=0.773, Glu r=0.596, sum of metabolites r=0.888, all p<0.0001). Therefore, the data for each individual metabolite for each animal, for each session were normalized on the basis of these metabolite-tissue concentration regression functions with the results expressed as standard deviations from an expected mean value of zero. Additionally, the ratio of NAA/Cr was determined on the absolute metabolite concentration data. For graphical display, grand-average spectra were computed separately for the thiamine-deficient and thiamine-treated groups with the spectra normalized to maximum peak height of Cr across all animals and all sessions.

(a) Five contiguous coronal slices (anterior, left to posterior, right) subtending the MRS voxel, which is displayed as the white rectangle on each slice. (b) Water peak of each of 16 echoes; the higher peaks are from the earlier echoes and the shorter ones from the later echoes. (c) Full bi-exponential T2 relaxation model fit (black), with tissue and CSF components (gray). Regression analyses assessed the influence of the amount of tissue excited in the PRESS box.
Statistical Analysis
Within- and between-group differences were examined with repeated measures analysis of variance (ANOVA) and follow-up t-tests or Scheffe tests (α=0.05, one-tailed for directional hypotheses). Because two rats in the pyrithiamine/alcohol condition died before the second post-treatment MRI, the analyses were performed twice, once with all 18 rats across two MRI sessions and again with the 16 survivors across three MRI sessions. Examination of the effects of prior alcohol exposure was inherent in the design of the present study.
RESULTS
Thiamine Deprivation Effect on Body Weight and Neurological Status
All pyrithiamine-treated animals lost weight (on average, 74 g (11.7%), range=9.3–12.4%), whereas the thiamine-treated controls gained an average of 13 g (2.3%) from the beginning to the end of the 14-day treatment (t(16)=13.78, p=0.0001). The pyrithiamine-treated animals showed only a modest and nonsignificant restitution of weight, on average, a 24 g increase by the last scan date (Figure 3). Post hoc analysis of the eight surviving pyrithiamine-treated rats divided on alcohol exposure history revealed that weight gain between MRI 2 and 3 occurred only for those without alcohol exposure (subgroup-by-session interaction F(1,6)=6.74, p=0.0409).

Mean±SEM of body weight at five times for each of the four groups: TW=thiamine+water pre-exposure group; TA=thiamine+alcohol pre-exposure group; PW=pyrithiamine+water pre-exposure group; and PA=pyrithiamine+alcohol pre-exposure group. TD=thiamine deficiency. Note that only the mean±SEM at the late post-TD time is based on the three surviving (of five initial) rats in that group.
In spite of weight loss, detectable brain lesions, and regardless of alcohol exposure, only half of the pyrithiamine-treated rats (three with prior alcohol exposure) displayed clinical signs of thiamine deficiency, including seizure-like episodes, lethargy, splayed paw placement, questionable blindness, failure to respond to stimulation, tremor, and inability to stand on hind paws. Some failed to eat and were treated with gavage of nutritional supplement until they resumed voluntary eating.
Structural MRI: Regional Brain Changes from Thiamine Deprivation
MRI signal intensity change
Pyrithiamine treatment produced striking signal intensity increases, clinically visible, in all animals on both early- and late-echo images. Irrespective of prior alcohol exposure, the post–pre image intensities were significantly greater in the pyrithiamine than thiamine groups for the thalamus, mammillary bodies, and inferior colliculi. A summary of these results is presented next, and the ANOVA values appear in Table 2. Group differences in these brain regions were not related to prior alcohol exposure by itself or in interaction with pyrithiamine treatment for any measure (early-echo or late-echo signal intensity and T2 relaxation time) in three-session omnibus analyses; therefore, primary analysis did not include alcohol as a grouping variable. None of the analyses revealed significant differences at baseline between the thiamine-deprived and thiamine-replete groups. Means±SEM of these measures for each region are presented in Figure 4 (thalamus), Figure 5 (T2 relaxation times for the three brain regions), Figure 6 (mammillary nuclei), and Figure 7 (inferior colliculi).

Left: Mean±SEM of early-echo and late-echo signal intensities at three experiment times for each of the four groups: TW=thiamine+water pre-exposure group; TA=thiamine+alcohol pre-exposure group; PW=pyrithiamine+water pre-exposure group; and PA=pyrithiamine+alcohol pre-exposure group. TD=thiamine deficiency. Right: Coronal images (top set of six images=early echo; bottom set of six images=late echo) displaying the enhanced signal intensity, reflective of pathology in the thalamus, from pre-TD to early post-TD and reduction of the signal intensity late post-TD, reflective of recovery, from the three experimental times for a pyrithiamine-treated rat and a thiamine-treated rat.

Mean±SEM of transverse relaxation time in three brain structures at three experiment times for each of the four groups: TW=thiamine+water pre-exposure group; TA=thiamine+alcohol pre-exposure group; PW=pyrithiamine+water pre-exposure group; and PA=pyrithiamine+alcohol pre-exposure group. TD=thiamine deficiency.

Left: Mean±SEM of early-echo and late-echo signal intensities at three experiment times for each of the four groups: TW=thiamine+water pre-exposure group; TA=thiamine+alcohol pre-exposure group; PW=pyrithiamine+water pre-exposure group; and PA=pyrithiamine+alcohol pre-exposure group. TD=thiamine deficiency. Right: Coronal images (top set of six images=early echo; bottom set of six images=late echo) displaying the enhanced signal intensity, reflective of pathology in the mammillary nuclei, from pre-TD to early post-TD and no reduction of the signal intensity late post-TD, reflective of lack of recovery, from the three experimental times for a pyrithiamine-treated rat and a thiamine-treated rat.

Left: Mean±SEM of early-echo and late-echo signal intensities at three experiment times for each of the four groups: TW=thiamine+water pre-exposure group; TA=thiamine+alcohol pre-exposure group; PW=pyrithiamine+water pre-exposure group; and PA=pyrithiamine+alcohol pre-exposure group. TD=thiamine deficiency. Right: Coronal images (top set of six images=early echo; bottom set of six images=late echo) displaying the enhanced signal intensity, reflective of pathology in the inferior colliculi, from pre-TD to early post-TD and reduction of the signal intensity late post-TD, reflective of recovery, from the three experimental times for a pyrithiamine-treated rat and a thiamine-treated rat.
Thalamus
The two-session and three-session ANOVAs revealed the same pattern for all MRI tissue measures, where the group effect and group-by-session interaction were significant. Follow-up t-tests indicated that in no cases did the pyrithiamine- and thiamine-treated groups differ at baseline, but there were significant group differences at session 2. Thus, the early-echo, late-echo, and T2 values in the thalamus of the pyrithiamine-treated group were disproportionately greater at MRI session 2 than those of the thiamine-treated group.
These differences persisted for the early echo, but not late echo or T2, at MRI session 3. Comparison of sessions 2–3, an index of recovery, revealed significant reduction in the signal hyperintensity for early echo (p=0.0002), late echo (p=0.0005), and T2 (p=0.007). Further analyses examined the effect of prior alcohol exposure on recovery in the pyrithiamine group and identified a subgroup-by-session interaction for T2 (F(1,6)=7.575, p=0.0332) and a trend for late-echo signal intensity change (F(1,6)=5.603, p=0.0557), suggesting attenuated recovery in the dually treated subgroup; thiamine-deficient animals with prior alcohol exposure showed less recovery.
Mammillary bodies
Group-by-session interactions were significant for the two- and three-session comparisons of the early-echo (p=0.0001 for both sessions) and late-echo data (two session p=0.03; three session p<0.01). By contrast, the T2 analyses failed to yield significant group effects or group-by-session interactions. Comparison of sessions 2–3 revealed no reduction in the signal hyperintensity for early echo, late echo, or T2, and no significant relation to alcohol history.
Inferior colliculus
As with the thalamus and mammillary bodies, group-by-session interactions were significant for the two-session and three-session comparisons of the early-echo (p=0.0001, both sessions) and late-echo data (two session p=0.0001; three session p=0.0003). The T2 estimates were significant for both the two-session (p=0.0012) and three-session (p<0.03) comparisons. Sessions 2 to 3 comparisons revealed significant reduction in the signal hyperintensity for early-echo (p=0.0001), late-echo (p=0.007), and T2 (p<0.05) without a significant alcohol history effect.
Change in brain structural size
Manual measurement revealed significant enlargement of the lateral ventricles in the pyrithiamine-treated compared with the thiamine-treated animals for the two-session (group-by-session interaction F(1,16)=5.04, p<0.04) and three-session (interaction F(2,28)=6.40, p<0.006) data, which was not related to alcohol exposure (Figure 8). The ventricles expanded further from sessions 2 to 3 in the thiamine-deficient group (F(1,14)=5.667, p=0.032), again with no change in the thiamine-treated group or significant alcohol history effect.

Left: Mean±SEM of volume of the lateral ventricles at three experiment times for each of the four groups: TW=thiamine+water pre-exposure group; TA=thiamine+alcohol pre-exposure group; PW=pyrithiamine+water pre-exposure group; and PA=pyrithiamine+alcohol pre-exposure group. TD=thiamine deficiency. Right: Coronal images displaying ventricular enlargement from pre-TD to early post-TD and lack of recovery late post-TD, from the three experimental times for a pyrithiamine-treated rat and a thiamine-treated rat.
Despite the significant alcohol effect (F(1,14)=4.82, p<0.05) on corpus callosum size measurable at baseline of the present experiment, thiamine deficiency did not cause further callosal shrinkage (two-session group-by-session interaction F(1,16)=0.08, p=0.78; three-session interaction F(2,28)=1.66, p=0.21). Analyses of the effect of prior alcohol exposure on recovery in the pyrithiamine-treated group identified a subgroup-by-session interaction (F(1,6)=3.719, p=0.0423), owing to growth in callosal size in the non-alcohol-exposed group. The cerebellum showed neither alcohol nor thiamine-depletion effects in either the two-session (F(1,16)=0.04, p=0.84) or three-session (F(2,28)=0.34, p=0.71) comparison.
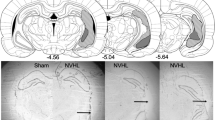
Post hoc whole brain t-test survey
After the initial region-of-interest analysis, a whole brain survey of parenchymal changes was conducted by computing t-tests on a voxel-by-voxel basis for early- and late-echo data across treatment groups and sessions using a p-value of <0.01 to define significant differences. As expected, both early- and late-echo data yielded prominent clusters of significant t-test differences in the same three structures noted in the region-of-interest analyses: thalamus (Figure 9), mammillary body nuclei (Figure 10), and inferior colliculi (Figure 11). Inspection of slices on which the hippocampus was present also revealed regions of apparent hyperintensity at the dorsal rim of the dorsal hippocampus and medial margins of the ventral hippocampus (Figures 9 and 10). Enlargement of the lateral ventricles was evident on late-echo data (not shown) as was enlargement of the fourth ventricle with no visible recovery (Figure 12), not detected in the region-of-interest analysis.

Top panel of five coronal images are based on early-echo data, and the bottom panel presents the late-echo data. The bottom two images in each panel show the outline of each image and the voxel-by-voxel t-test differences meeting a p<0.01 statistical significance criterion between pre-TD and early post-TD images (left) and between early and late post-TD images.

Top panel of five coronal images are based on early-echo data, and the bottom panel presents the late-echo data. The bottom two images in each panel show the outline of each image and the voxel-by-voxel t-test differences meeting a p<0.01 statistical significance criterion between pre-TD and early post-TD images (left) and between early and late post-TD images.

Top panel of five coronal images are based on early-echo data, and the bottom panel presents the late-echo data. The bottom two images in each panel show the outline of each image and the voxel-by-voxel t-test differences meeting a p<0.01 statistical significance criterion between pre-TD and early post-TD images (left) and between early and late post-TD images.

Five coronal images are based on late-echo data. The bottom two images show the outline of each image and the voxel-by-voxel t-test differences meeting a p<0.01 statistical significance criterion between pre-TD and early post-TD images (left) and between early and late post-TD images.
Proton MRS: Effect of Thiamine Deprivation on Major Proton Metabolites
Voxel placement for MRS metabolite quantification subtended the thalamus. Tissue content of each voxel at each scanning session was estimated with a bi-exponential T2 fit of the unsuppressed 2D J water signal. The tissue T2 of this voxel was examined across the three MRS sessions, and, consistent with the structural region-of-interest analysis, group-by-session ANOVA revealed a significant interaction (F(2,28)=3.39, p=0.048) across the three scanning sessions (initial increase and then return) and no effect of alcohol exposure on primary, three-session analyses (Figure 13).

Overlapping grand average MRS spectra over the three experiment collection times for the pyrithiamine-treated rats (top set of three spectra) and the thiamine-treated rats (bottom set of three spectra). The NAA peaks noted in the ovals are presented again as sequential peaks for a clearer display of the treatment effects.
Separate group-by-session ANOVAs were then conducted for each tissue concentration corrected metabolite quantified: NAA, Cr, Cr2, Cho, and Glu (Figure 14). The only metabolite showing either a main effect of group or an interaction was NAA (three sessions: group F(1,14)=12.257, p=0.0035; interaction F(2,28)=3.767, p=0.0356). Follow-up ANOVAs yielded group-by-session interactions, indicating a 2 SD decline from baseline to the early post-thiamine-deficient MRS session (F(1,16)=9.52, p=0.0071) followed by a trend for recovery at the later post-treatment session (F(1,14)=4.41, p=0.0543). Despite the recovery, a 1.58 SD deficit in NAA endured in the thiamine-deficient group and was significantly lower than that of the controls (t(14)=2.153, p<0.05).

Mean±SEM of the five metabolites quantified and the NAA/Cr ratio for each of the three experimental times for the thiamine-treated rats (open circles) and pyrithiamine-treated rats (closed circles).
The analyses based on NAA concentration were repeated for the ratio of NAA/Cr, a metric commonly used in clinical studies. The pattern of results was the same as with the concentration values, where the group effect (F(1,14)=14.421, p=0.002) and interaction (F(2,28)=3.554, p=0.0421) were significant for the three-session ANOVA (Figure 14). Two-session ANOVAs further supported an initial decline (F(1,16)=5.45, p=0.0329) followed by significant recovery from sessions 2 to 3 (F(1,14)=7.04, p=0.0189). Analyses of the effect of prior alcohol exposure on recovery in the pyrithiamine-treated group identified a subgroup-by-session interaction for NAA/Cr (F(1,6)=7.226, p=0.0361), suggesting disproportionately greater improvement in the non-alcohol-exposed rats (Figure 15). Expression of the Cho concentrations as ratios of NAA or Cr yielded no significant group differences at any session. Neither were there any significant treatment effects on Glu, which is well resolved with 2D J spectroscopy.

Mean±SEM of the MRS-derived NAA/Cr ratio at three experiment times for each of the four groups: TW=thiamine+water pre-exposure group; TA=thiamine+alcohol pre-exposure group; PW=pyrithiamine+water pre-exposure group; and PA=pyrithiamine+alcohol pre-exposure group. TD=thiamine deficiency.
Clinical Status and Brain Measures
Five pyrithiamine-treated animals manifested significant clinical signs of thiamine deficiency and five (three alcohol-naïve, two alcohol-exposed) did not, despite increases in thalamic signal intensity in all 10 rats and decline in NAA in nine of the 10. Repeated-measures ANOVAs within the pyrithiamine-treated animals revealed significant differences in body weight and brain changes as a function of presence or absence of clinical signs. Rats that manifested clinical signs lost more weight and had less recovery of body weight (p<0.01) and had more signal intensity increase and less recovery toward baseline values for both early-echo (p<0.02) and late-echo (p<0.01) data than those without clinical signs. NAA concentration also declined more from baseline to first post-treatment MRS in rats with clinical signs than those without clinical signs (p<0.02).
DISCUSSION
Using in vivo quantitative MRI and MRS in a longitudinal design, we were able to support, in large part, the hypotheses posed. Firstly, we were successful in producing the signature lesions of WE, involving the thalamus, inferior colliculi, and mammillary body nuclei and dilatation of the lateral ventricles, with a single bout of thiamine deficiency, using daily i.p. pyrithiamine injections coupled with a thiamine-depleted diet for 2 weeks. A voxel-based statistical survey also identified fourth ventricular enlargement and regions of the dorsal and ventral hippocampus as additional areas of thiamine deficiency-induced hyperintense tissue. These lesions and ventricular expansions were visible with early post-treatment, structural MRI, acquired 3 weeks after re-instatement of a thiamine-enriched diet and 4–6 days after a single i.p. bolus of thiamine. Secondly, when examined later post-treatment (35 days after reintroduction of dietary thiamine), tissue recovery, quantified as reduction in regions of hyperintense signal, was significant in the thalamus and inferior colliculus but not in the mammillary nuclei, hippocampus, or ventricular system. Concurrent biochemical assessment of the thalamus using MRS provided evidence for cell damage, indexed by a significant decline in NAA, followed by a significant rise, indicative of recovery. Thirdly, both MRI and MRS data provided evidence of an untoward effect of prior chronic alcohol exposure in thiamine-deficient rats in the form of attenuated recovery of the thalamus. Moreover, two rats died before the last scanning session, and both had received both exposures, alcohol and pyrithiamine (cf, Zimitat et al, 1990). Finally, although we did not observe an exacerbation of the thiamine deprivation effect on alcohol-related reduction in corpus callosum size, rats with both exposure histories showed attenuation of normal callosal growth, which was evident in comparison with thiamine-treated rats during the later recovery period.
Pattern of Neuroradiological Signs Induced by Thiamine Deficiency
Animal models of the brain lesions marking the thiamine deficiency syndrome have typically revealed subsets of the lesions noted in humans (reviewed by Martin et al, 2003; Witt, 1985), involving the thalamus, superior and inferior colliculi, and hypothalamic nuclei. Differences among studies have been attributed to differences in the method of thiamine deficiency induction (diet alone vs pyrithiamine), number of thiamine deficiency bouts, severity of thiamine deficiency sequelae, time of examination with relation to thiamine deficiency manipulation, and species differences. A further variable is the limitation in identifying lesions in widespread brain regions post mortem because of the labor-intensive examination required for whole brain analysis and the limitations imposed on exploratory analysis by tissue preparation and cutting. Magnetic resonance neuroimaging study has its own challenges, but can provide repeated, longitudinal in vivo high-resolution surveys of the whole brain and its structures that can be analyzed with a variety of approaches, including metabolite analyses of tissue, and longitudinal examinations of before and after treatment effects.
To date, the few MRI studies of thiamine deficiency in animal models have all used rats. The first set of studies revealed ventricular enlargement, confirmed histologically, in Sprague–Dawley rats following a thiamine-deficient diet that continued until their baseline body weight was reduced by 40% (about 6 weeks) (Acara et al, 1995; Pentney et al, 1993). Parenchymal changes, noted as increased T2, also occurred and were indicative of inter- and extracellular fluid accumulation. Although these radiological signs concurred with human clinical radiological signs of WE, the resolution and registration of the images in that study precluded localizing the hyperintensities to specific brain structures. Another longitudinal MRI experiment conducted with female wild-type Wistar rats used a multi-echo image acquisition sequence permitting calculation of transverse relaxation time (Jordan et al, 1998). Despite limited brain coverage, this study revealed bilaterally distributed hyperintense signal on T2-weighted images, indicative of tissue pathology in thalamus, hypothalamus, collicular bodies, and hippocampus. Further, glucose administration to thiamine-deficient rats exacerbated lesions in the ventral hippocampus that were only marginally visible without the glucose challenge and may be related to glucose-induced impairment of the blood–brain barrier (Zelaya et al, 1995). The glucose-enhanced hippocampal hyperintensity endured for at least a month, and the authors concluded that the hippocampal abnormality was as persistent and reliable as the well-established thalamic lesion marking WE. Consistent with that study, we observed with whole-brain subtraction images prominent hyperintense margins of the dorsal and ventral hippocampus with little evidence for recovery.
Neurochemical Basis of Tissue Recovery
MR spectroscopy acquired concurrently with the structural brain data subtended the thalamus and permitted assessment of thalamic tissue metabolites, particularly with respect to neuronal integrity measured with NAA, cell membrane metabolism with Cho, and energy production with Cr (Petroff et al, 1991, 1995). The only major visible proton metabolite that changed with thiamine manipulation was NAA; none of the other peaks (Cr, Cho, or Glu) varied across test conditions. NAA, whether expressed as an absolute value or as a ratio of Cr, declined markedly from baseline to early post-pyrithiamine treatment and showed significant yet incomplete recovery from early to late post-pyrithiamine treatment. The spectroscopy results confirm the MRI findings of lesion development and partial resolution and extend these findings to indicate that initial compromise of neuronal integrity was substantially, albeit incompletely, reversed with thiamine treatment.
The selective effect of thiamine deficiency on neuronal integrity, that is, the NAA peak, observed herein with selectively bred rats of Wistar stock contrasts with prior MRS studies of thiamine deficiency conducted in Sprague–Dawley rats (Lee et al, 2001, 1995). Both of those two longitudinal studies found a significant decline in the Cho/NAA ratio soon after a regimen of pyrithiamine injections, followed by dose-dependent recovery with thiamine replenishment. The later study (Lee et al, 2001) confirmed the in vivo results with in vitro analysis and concluded that a determinant of the neuroanatomical lesions of WE relates to a deficit in Cho compounds. To address the discrepancy between the results of the Lee et al (2001, 1995) studies and the present study, group differences were re-examined using the Cho/NAA ratio but no differences related to treatment were detected. Whether the difference in study results is related to prior alcohol exposure or differences in experimental protocols, scanning parameters, or rat strains remains unresolved.
The MRS voxel used herein was limited to the thalamus and adjacent subcortical tissue. Surely, the cerebellum warrants closer MRS examination, as has been conducted in humans (Martin et al, 1995, 1992; Seitz et al, 1999), particularly of Cr and glutamate (Butterworth, 1993). Recently, we (Mayer et al, 2006) used in vivo CT-PRESS with a 0.2 ml voxel to quantitate regional differences in metabolite ratios and revealed that signal intensities of NAA, Cho, Glu, and Glx (relative to tCr) were significantly lower in the cerebellum than the striatum of the Wistar rat brain. Like 2D J, the CT-PRESS technique also provides the enhanced signal separation necessary to reliably measure J-coupled resonances, such as glutamate and myo-inositol (a marker of glial activity), in the rat brain at 3 T. Future studies using this approach may provide in vivo measures of a wide array of regionally focal proton metabolites that may serve to identify mechanisms of neuronal destruction and recovery (cf, Langlais et al, 1988).
Interaction of Thiamine Deficiency and Alcohol Exposure
One objective of our study was to test whether prior voluntary consumption of high doses of alcohol presented a vulnerability to exacerbated structural damage by thiamine deficiency. To address this potential interaction, we first considered the effect of long-term alcohol consumption on brain microstructure and morphology in rodents (cf, Pfefferbaum et al, 2006), which varies markedly with length of exposure. Upwards of 5 months of drinking ∼9% alcohol has been reported to produce substantial neuropathological changes, namely, decreased spine density and neuronal loss in the hippocampus and shrinkage of the granular cell layers notable in the superior vermis (Riley and Walker, 1978). Large amounts of dietary alcohol can also affect the integrity of the myelin of white matter fibers (eg, Harper and Kril, 1988; Wiggins et al, 1988). A histological study (Irle and Markowitsch, 1983) examined three groups of Sprague–Dawley rats: an alcohol-only group, which for 20 months had available 30% alcohol in water as the only liquid; a thiamine-deficient group, which for 3–4 weeks had no thiamine in the diet; and a control group. The clearest damage in the alcohol group occurred in the dentate gyrus of the dorsal hippocampus, neurons of the mammillary nuclei, and shrinkage of the granular layer and of pyramidal cells in the dorsal vermis. Additional structures showing pathology in the alcohol group were periaqueductal gray matter, collicular bodies, locus coeruleus, olfactory tubercle, and prepiriform cortex. Structures consistently affected in the thiamine-deficient group were the ventricles, mammillary nuclei, and cerebellar cortex (superior vermis). Langlais and Zhang (1997) have reported evidence for a temporal accruing of thiamine deficiency-induced neuropathology. Specifically, Sprague–Dawley rats were administered pyrithiamine for 12–13 days or for 14–15 days in a single bout. Histology of the shorter-duration thiamine deficiency group revealed widely spread axonal degeneration that included large commissural tracts, whereas only the longer-treated group exhibited degenerating axons in the cortex and hippocampus and neuronal loss in mammillary bodies and medial thalamus. Although such microstructural abnormalities are below detection with MRI, we recently observed in adult P rats that selective brain structures, notably, the corpus callosum, cerebellum, and hippocampus, grow significantly over 1 year in adulthood (Sullivan et al, 2006) and that voluntary consumption of large amounts of alcohol in a three-bottle choice regime (15% alcohol, 30% alcohol, and water) attenuated brain growth, particularly of the corpus callosum (Pfefferbaum et al, 2006).
In other work testing whether thiamine deficiency exacerbates the untoward effects of excessive alcohol consumption, Zimitat et al (1990) examined wild-type Wistar rats either with or without ad lib 15% alcohol in water or thiamine-deficient diet. Although neuropathological investigation failed to yield a compounded effect of alcohol consumption and thiamine deficiency, the onset of the behavioral sequelae developed earlier in the dually treated rats. We made a similar observation in the present experiment, in that the only rats that died prematurely exhibited severe neurological sequelae of pyrithiamine treatment, required post-pyrithiamine nutritional supplement by gavage, and had had prior alcohol exposure. In addition, we noted modest alcohol–thiamine-deficiency interactions, such that dually treated rats showed modestly attenuated structural and neuronal recovery in the thalamus and arrested growth of the corpus callosum.
In our previous experiment (Pfefferbaum et al, 2006), the alcohol-exposed rats gained less weight than their water control counterparts. Body weight loss, as commonly occurs in alcoholic humans, or lack of normal weight gain, as occurred in our alcohol-consuming rats, may signal a nutritional–alcohol interaction, putting alcohol-exposed animals or humans at risk for nutritional deficiencies (eg, Harper and Butterworth, 1997; Martin et al, 2003; Zimatkin and Zimatkina, 1996) and alcoholism-related brain dysmorphology (eg, Pfefferbaum et al, 2004b). All rats, regardless of alcohol history, were obese at start of the current experiment and thus even the alcohol-drinking rats had eaten vitamin-fortified chow given them and apparently had adequate nutrition while drinking, which may have mitigated potential brain damage (cf, Harper and Butterworth, 1997; Langlais and Savage, 1995; Martin et al, 2003; Pentney et al, 1993; Zimatkin and Zimatkina, 1996). Compromise of thiamine absorption with alcohol dependence and surely with pyrithiamine treatment is one recognized mechanism promoting brain structural pathology (for review, Lieber, 2003b).
Cerebellar damage is a common outcome of thiamine deficiency and chronic alcohol ingestion. An in vitro study of cerebellar cultures of Sprague–Dawley rat pups revealed that cerebellar pathology is more related to thiamine deficiency than to alcohol withdrawal per se in alcohol dependency (Mulholland et al, 2005). Additionally, the damage may have a serotoninergic substrate (cf, Bloom et al, 1972; Halliday et al, 1995). Another in vitro study reported decreased serotonin (5-HT) uptake in cerebellar synaptosomes taken from Sprague–Dawley rats made thiamine deficient either by diet or pyrithiamine (Plaitakis et al, 1978). It is possible that reduced 5-HT activity in selectively bred P, compared with selectively bred alcohol non-preferring, rats (McBride et al, 1991; Zhou et al, 1995) may have protected the rats in the present study from notable cerebellar shrinkage.
Conclusion
Differences in regional patterns of affected brain structures and identification of alcohol and thiamine deficiency compounded effects may help distinguish alcohol-associated effects from thiamine deficiency effects on brain structure and function. The findings of the present study indicated that the regions clearly affected by thiamine deficiency in P rats were the thalamus, mammillary nuclei, inferior colliculi, lateral and fourth ventricles, and hippocampus. Some of these structures, including the hippocampus, may have a special vulnerability to neurotoxic agents, including alcohol, and may also be able to regenerate when these toxins are removed (cf, Nixon and Crews, 2002, 2004). The parenchymal and ventricular changes observed with thiamine manipulation in the present study concur with human radiological signs of WE. A limitation of the present study is the lack of formal behavioral testing of the study animals. Nonetheless, the enduring macrostructural and neurochemical abnormalities involving critical nodes of the Papez (1937) circuit carry liabilities for development of amnesia and incomplete recovery of nonmnemonic cognitive and motor functions subserved by the affected neural systems (cf, Dixon and Harper, 2001). Neuroimaging using animal models of alcoholism, with the P rat meeting criteria proposed for an animal model of alcoholism (cf, McBride and Li, 1998; Murphy et al, 2002; Bell et al, 2005) and thiamine deficiency, provides a unique approach to longitudinal investigation of these neuronal and behavioral systems.
References
Acara M, Alletto JJ, Dlugos C, Pentney R (1995). Small animal MRI. Alcohol Health Res World 4: 321–324.
Adalsteinsson E, Hurd RE, Mayer D, Sailasuta N, Sullivan EV, Pfefferbaum A (2004). In vivo 2D J-resolved magnetic resonance spectroscopy of rat brain with a 3T clinical human scanner. Neuroimage 22: 381–386.
Adalsteinsson E, Sullivan E, Kleinhans N, Spielman D, Pfefferbaum A (2000). Longitudinal decline of the neuronal marker NAA in Alzhiemer's disease as measured by 1H magnetic resonance spectroscopic imaging (abs). Presented at Alzheimer's Imaging Consortium, Washington, DC, July 8.
Adalsteinsson E, Sullivan EV, Mayer D, Pfefferbaum A (2006). In vivo quantification of ethanol kinetics in rat brain. Neuropsychopharmacology [Epub ahead of print January 11].
Agartz I, Momenan R, Rawlings RR, Kerich MJ, Hommer DW (1999). Hippocampal volume in patients with alcohol dependence. Arch Gen Psychiatry 56: 356–363.
Baker K, Harding A, Halliday G, Kril J, Harper C (1999). Neuronal loss in functional zones of the cerebellum of chronic alcoholics with and without Wernicke's encephalopathy. Neuroscience 91: 429–438.
Bell RL, Rodd ZA, Murphy JM, McBride WJ (2005). Use of selectively bred alcohol-preferring rats to study alcohol abuse, relapse and craving. In: Preedy VR, Watson RR (eds). Comprehensive Handbook of Alcohol Related Pathology, Vol 3. New York: Academic Press, Elsevier Science. pp 1515–1533.
Bender L, Schilder P (1933). Encphalopathia alcoholica: polioencephalitis haemorrhagica superior of Wernicke. Arch Neurol Psychiatry 29: 990–1053.
Bendszus M, Weijers HG, Wiesbeck G, Warmuth-Metz M, Bartsch AJ, Engels S et al (2001). Sequential MR imaging and proton MR spectroscopy in patients who underwent recent detoxification for chronic alcoholism: correlation with clinical and neuropsychological data. Am J Neuroradiol 22: 1926–1932.
Blansjaar B, Vielvoye G, van Dijk J, Rijnders R (1992). Similar brain lesions in alcoholics and Korsakoff patients: MRI, psychometric and clinical findings. Clin Neurol Neurosurg 93: 197–203.
Bloom FE, Hoffer BJ, Siggins GR, Barker JL, Nicoll RA (1972). Effects of serotonin on central neurons: microiontophoretic administration. Fed Proc 31: 97–106.
Butters N, Cermak LS (1980). Alcoholic Korsakoff's Syndrome: An Information Processing Approach to Amnesia. Academic Press Inc.: New York.
Butterworth RF (1993). Pathophysiology of cerebellar dysfunction in the Wernicke–Korsakoff syndrome. Can J Neurol Sci 20: S123–S126.
Caine D, Halliday GM, Kril JJ, Harper CG (1997). Operational criteria for the classification of chronic alcoholics: identification of Wernicke's encephalopathy. J Neurol Neurosurg Psychiatry 62: 51–60.
Cardenas VA, Studholme C, Meyerhoff DJ, Song E, Weiner MW (2005). Chronic active heavy drinking and family history of problem drinking modulate regional brain tissue volumes. Psychiatry Res 138: 115–130.
Carlen PL, Wortzman G, Holgate RC, Wilkinson DA, Rankin JG (1978). Reversible cerebral atrophy in recently abstinent chronic alcoholics measured by computed tomography scans. Science 200: 1076–1078.
Charness ME (1993). Brain lesions in alcoholics. Alcohol Clin Exp Res 17: 2–11.
Charness ME (1999). Intracranial voyeurism: revealing the mammillary bodies in alcoholism. Alcohol Clin Exp Res 23: 1941–1944.
Charness ME, DeLaPaz RL (1987). Mammillary body atrophy in Wernicke's encephalopathy: antemortem identification using magnetic resonance imaging. Ann Neurol 22: 595–600.
Chu K, Kang DW, Kim HJ, Lee YS, Park SH (2002). Diffusion-weighted imaging abnormalities in wernicke encephalopathy: reversible cytotoxic edema? Arch Neurol 59: 123–127.
Ciccia RM, Langlais PJ (2000). An examination of the synergistic interaction of ethanol and thiamine deficiency in the development of neurological signs and long-term cognitive and memory impairments. Alcohol Clin Exp Res 24: 622–634.
Davila MD, Shear PK, Lane B, Sullivan EV, Pfefferbaum A (1994). Mammillary body and cerebellar shrinkage in chronic alcoholics: an MRI and neuropsychological study. Neuropsychology 8: 433–444.
De Bellis MD, Narasimhan A, Thatcher DL, Keshavan MS, Soloff P, Clark DB (2005). Prefrontal cortex, thalamus, and cerebellar volumes in adolescents and young adults with adolescent-onset alcohol use disorders and comorbid mental disorders. Alcohol Clin Exp Res 29: 1590–1600.
Dixon G, Harper CG (2001). Quantitative analysis of glutamic acid decarboxylase-immunoreactive neurons in the anterior thalamus of the human brain. Brain Res 923: 39–44.
Dlugos CA, Pentney RJ (1997). Morphometric evidence that the total number of synapses on Purkinje neurons of old F344 rats is reduced after long-term ethanol treatment and restored to control levels after recovery. Alcohol Alcoholism 32: 161–172.
Doraiswamy PM, Massey EW, Enright K, Palese VJ, Lamonica D, Boyko O (1994). Wernicke–Korsakoff syndrome caused by psychogenic food refusal – MR findings. Am J Neuroradiol 15: 594–596.
Dreher W, Leibfritz D (1995). On the use of two-dimensional-J NMR measurements for in vivo proton MRS: measurement of homonuclear decoupled spectra without the need for short echo times. Magn Reson Med 34: 331–337.
Durazzo TC, Gazdzinski S, Banys P, Meyerhoff DJ (2004). Cigarette smoking exacerbates chronic alcohol-induced brain damage: a preliminary metabolite imaging study. Alcoholism Clin Exp Res 28: 1849–1860.
Eckardt M, File S, Gessa G, Grant K, Guerri C, Hoffman P et al (1998). Effects of moderate alcohol consumption on the central nervous system. Alcohol Clin Exp Res 22: 351–359.
Ende G, Welzel H, Walter S, Weber-Fahr W, Diehl A, Hermann D et al (2005). Monitoring the effects of chronic alcohol consumption and abstinence on brain metabolism: a longitudinal proton magnetic resonance spectroscopy study. Biol Psychiatry 58: 974–980.
Fein G, Bachman L, Fisher S, Davenport L (1990). Cognitive impairments in abstinent alcoholics. Western J Med 152: 531–537.
Fein G, Di Sclafani V, Cardenas VA, Goldmann H, Tolou-Shams M, Meyerhoff DJ (2002). Cortical gray matter loss in treatment-naive alcohol dependent individuals. Alcohol Clin Exp Res 26: 558–564.
Fein G, Meyerhoff D, Di Sclafani V, Ezekiel F, Poole N, MacKay S et al (1994). H magnetic resonance spectroscopic imaging separates neuronal from glial changes in alcohol-related brain atrophy. In: Lancaster F (ed). Alcohol and Glial Cells NIAAA Research Monograph # 27. US Government Printing Office: Bethesda, MD. pp 227–241.
Gazdzinski S, Durazzo TC, Meyerhoff DJ (2005). Temporal dynamics and determinants of whole brain tissue volume changes during recovery from alcohol dependence. Drug Alcohol Depend 78: 263–273.
Haase A, Frahm J, Hanicke W, Matthaei D (1985). H NMR chemical shift selective (CHESS) imaging. Phys Med Biol 30: 341–344.
Halliday G, Baker K, Harper C (1995). Serotonin and alcohol-related brain damage. Metab Brain Dis 10: 25–30.
Harper C, Butterworth R (1997). Nutritional and metabolic disorders. In: Graham DI, Lantos PL (eds). Greenfield's Neuropathology. London: Arnold. pp 601–642.
Harper C, Fornes P, Duyckaerts C, Lecomte D, Hauw JJ (1995). An international perspective on the prevalence of the Wernicke–Korsakoff syndrome. Metab Brain Dis 10: 17–24.
Harper CG, Kril JJ (1988). Corpus callosal thickness in alcoholics. Br J Addict 83: 577–580.
Harper CG, Kril JJ (1990). Neuropathology of alcoholism. Alcohol Alcohol 25: 207–216.
Harper CG, Sheedy DL, Lara AI, Garrick TM, Hilton JM, Raisanen J (1998). Prevalence of Wernicke–Korsakoff syndrome in Australia: has thiamine fortification made a difference? Med J Aust 168: 542–545.
Hurd RE (2000). Magnetic resonance spectroscopic imaging having reduced parasitic side band signals. United States patent no. 6,069,478, May 30, 2000.
Hurd RE, Gurr D, Sailasuta N (1998). Proton spectroscopy without water suppression: the oversampled J-resolved experiment. Magn Reson Med 40: 343–347.
Irle E, Markowitsch HJ (1983). Widespread neuroanatomical damage and learning deficits following chronic alcohol consumption or vitamin-B1 (thiamine) deficiency in rats. Behav Brain Res 9: 277–294.
Jagannathan NR, Desai NG, Raghunathan P (1996). Brain metabolite changes in alcoholism: an in vivo proton magnetic resonance spectroscopy (MRS) study. Magn Reson Imaging 14: 553–557.
Jernigan TL, Butters N, DiTraglia G, Schafer K, Smith T, Irwin M et al (1991). Reduced cerebral grey matter observed in alcoholics using magnetic resonance imaging. Alcohol Clin Exp Res 15: 418–427.
Jolliffe N, Wortis H, Fein HD (1941). The Wernicke syndrome. Arch Neurol Psychiatry 46: 569–597.
Jordan LR, Zelaya FO, Rose SE, Bower AJ, Galloway G, Wholohan T et al (1998). Changes in the hippocampus induced by glucose in thiamine deficient rats detected by MRI. Brain Res 791: 347–351.
Kopelman MD (1995). The Korsakoff syndrome. Br J Psychiatry 166: 154–173.
Kril JJ, Halliday GM, Svoboda MD, Cartwright H (1997). The cerebral cortex is damaged in chronic alcoholics. Neuroscience 79: 983–998.
Langlais PJ, Mair RG, Anderson CD, McEntee WJ (1988). Long-lasting changes in regional brain amino acids and monoamines in recovered pyrithiamine treated rats. Neurochem Res 13: 1199–1206.
Langlais PJ, Savage LM (1995). Thiamine deficiency in rats produces cognitive and memory deficits on spatial tasks that correlate with tissue loss in diencephalon, cortex and white matter. Behav Brain Res 68: 75–89.
Langlais PJ, Zhang SX (1997). Cortical and subcortical white matter damage without Wernicke's encephalopathy after recovery from thiamine deficiency in the rat. Alcohol Clin Exp Res 21: 434–443.
Lee H, Holburn GE, Price RR (2001). In vivo and in vitro proton NMR spectroscopic studies of thiamine-deficient rat brains. J Magn Reson Imaging 13: 163–166.
Lee H, Holburn GH, Price RR (2003). Proton MR spectroscopic studies of chronic alcohol exposure on the rat brain. J Magn Reson Imaging 18: 147–151.
Lee H, Tarter J, Holbourn GE, Price RR, Weinstein DD, Martin PR (1995). In vivo localized proton NMR spectroscopy of thiamine-deficient rat brain. Magn Reson Med 34: 313–318.
Li TK, Lumeng L, McBride WJ, Waller MB (1979). Progress toward a voluntary oral consumption model of alcoholism. Drug Alcohol Depend 4: 45–60.
Lieber CS (2003a). Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health 27: 220–231.
Lieber CS (2003b). Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health 27: 220–231.
Lu W, Jaatinen P, Rintala J, Sarviharju M, Kiianmaa K, Hervonen A (1997). Effects of lifelong ethanol consumption on rat locus coeruleus. Alcohol Alcohol 32: 463–470.
Mann K, Gunther A, Stetter F, Ackermann K (1999). Rapid recovery from cognitive deficits in abstinent alcoholics: a controlled test–retest study. Alcohol Alcohol 34: 567–574.
Martin PR, Gibbs SJ, Nimmerrichter AA, Riddle WR, Welch LW, Willcott MR (1995). Brain proton magnetic resonance spectroscopy studies in recently abstinent alcoholics. Alcohol Clin Exp Res 19: 1078–1082.
Martin PR, Rio D, Adinoff B, Johnson JL, Bisserbe J-C, Rawlings RR et al (1992). Regional cerebral glucose utilization in chronic organic mental disorders associated with alcoholism. J Neuropsychiatry Clin Neurosci 4: 159–167.
Martin PR, Singleton CK, Hiller-Sturmhofel S (2003). The role of thiamine deficiency in alcoholic brain disease. Alcohol Res Health 27: 134–142.
Mason GF, Petrakis IL, de Graaf RA, Gueorguieva R, Guidone E, Coric V et al (2006). Cortical gamma-aminobutyric acid levels and the recovery from ethanol dependence: preliminary evidence of modification by cigarette smoking. Biol Psychiatry 59: 85–93.
Mayer D, Zahr NM, Sullivan EV, Pfefferbaum A (2006). Differences in cerebellar and striatal metabolite ratios in rat brain detected in vivo with optimized CT-PRESS at 3T (abs). Presented at International Society for Magnetic Resonance in Medicine, Seattle, WA, May.
McBride WJ, Li TK (1998). Animal models of alcoholism: neurobiology of high alcohol-drinking behavior in rodents. Crit Rev Neurobiol 12: 339–369.
McBride WJ, Murphy JM, Gatto GJ, Levy AD, Lumeng L, Li TK (1991). Serotonin and dopamine systems regulating alcohol intake. Alcohol Alcohol 1: 411–416.
Meyerhoff DJ, Blumenfeld R, Truran D, Lindgren J, Flenniken D, Cardenas V et al (2004). Effects of heavy drinking, binge drinking, and family history of alcoholism on regional brain metabolites. Alcohol Clin Exp Res 28: 650–661.
Mulholland PJ, Self RL, Stepanyan TD, Little HJ, Littleton JM, Prendergast MA (2005). Thiamine deficiency in the pathogenesis of chronic ethanol-associated cerebellar damage in vitro. Neuroscience 135: 1129–1139.
Murata T, Fujito T, Kimura H, Omori M, Itoh H, Wada Y (2001). Serial MRI and (1)H-MRS of Wernicke's encephalopathy: report of a case with remarkable cerebellar lesions on MRI. Psychiatry Res 108: 49–55.
Murphy JM, Stewart RB, Bell RL, Badia-Elder NE, Carr LG, McBride WJ et al (2002). Phenotypic and genotypic characterization of the Indiana University rat lines selectively bred for high and low alcohol preference. Behavior Genetics 32: 363–388.
Nixon K, Crews FT (2002). Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. J Neurochem 83: 1087–1093.
Nixon K, Crews FT (2004). Temporally specific burst in cell proliferation increases hippocampal neurogenesis in protracted abstinence from alcohol. J Neurosci 24: 9714–9722.
O'Neill J, Cardenas VA, Meyerhoff DJ (2001). Effects of abstinence on the brain: quantitative magnetic resonance imaging and magnetic resonance spectroscopic imaging in chronic alcohol abuse. Alcohol Clin Exp Res 25: 1673–1682.
Papez JW (1937). A proposed mechanism of emotion. Arch Neurol 38: 725–743.
Parks MH, Dawant BM, Riddle WR, Hartmann SL, Dietrich MS, Nickel MK et al (2002). Longitudinal brain metabolic characterization of chronic alcoholics with proton magnetic resonance spectroscopy. Alcohol Clin Exp Res 26: 1368–1380.
Parsons O (1993). Impaired neuropsychological cognitive functioning in sober alcoholics. In: Hunt WA, Nixon SJ (eds). Alcohol Induced Brain Damage, NIAAA Research Monograph No. 22 National Institutes of Health: Rockville, MD. pp 173–194.
Pentney RJ, Alletto JJ, Acara MA, Dlugos CA, Fiel RJ (1993). Small animal magnetic resonance imaging: a means of studying the development of structural pathologies in the rat brain. Alcohol Clin Exp Res 17: 1301–1308.
Pentney RJ, Dlugos CA (2000). Cerebellar Purkinje neurons with altered terminal dendritic segments are present in all lobules of the cerebellar vermis of ageing, ethanol-treated F344 rats. Alcohol Alcohol 35: 35–43.
Pentney RJ, Quackenbush LJ (1990). Dendritic hypertrophy in Purkinje neurons of old Fischer 344 rats after long-term ethanol treatment. Alcohol Clin Exp Res 14: 878–886.
Petroff O, Ogino T, Alger J, Prichard J (1991). Regional variation in metabolite concentration and the effect of post-mortem autolysis: a magnetic resonance spectroscopy study of brain biopsy material. Society of Magnetic Resonance in Medicine Conference Abstracts Vol 61.
Petroff OAC, Pleban LA, Spencer DD (1995). Symbiosis between in vivo and in vitro NMR spectroscopy: the creatine, N-acetylaspartate, glutamate and GABA content of the epileptic human brain. Magn Reson Imaging 13: 1197–1211.
Pfefferbaum A, Adalsteinsson E, Sood R, Mayer D, Bell RL, McBride WJ et al (2006). Part II: longitudinal brain MRI study of the alcohol-preferring (P) rat: effects of voluntary chronic alcohol consumption. Alcohol Clin Exp Res (in press).
Pfefferbaum A, Adalsteinsson E, Spielman D, Sullivan EV, Lim KO (1999a). In vivo brain concentrations of N-acetyl compounds, creatine and choline in Alzheimer's disease. Arch Gen Psychiatry 56: 185–192.
Pfefferbaum A, Adalsteinsson E, Spielman D, Sullivan EV, Lim KO (1999b). In vivo spectroscopic quantification of the N-acetyl moiety, creatine and choline from large volumes of gray and white matter: effects of normal aging. Magn Reson Med 41: 276–284.
Pfefferbaum A, Adalsteinsson E, Sullivan EV (2004a). In vivo structural imaging of the rat brain with a 3T clinical human scanner. J Magn Reson Imaging 20: 779–785.
Pfefferbaum A, Adalsteinsson E, Sullivan EV (2005). Cortical NAA deficits in HIV infection without dementia: influence of alcoholism comorbidity. Neuropsychopharmacology 30: 1392–1399.
Pfefferbaum A, Lim KO, Zipursky RB, Mathalon DH, Lane B, Ha CN et al (1992). Brain gray and white matter volume loss accelerates with aging in chronic alcoholics: a quantitative MRI study. Alcohol Clin Exp Res 16: 1078–1089.
Pfefferbaum A, Rosenbloom MJ, Serventi KL, Sullivan EV (2004b). Brain volumes, RBC status, and hepatic function in alcoholics after 1 and 4 weeks of sobriety: predictors of outcome. Am J Psychiatry 161: 1190–1196.
Pfefferbaum A, Sullivan EV, Mathalon DH, Lim KO (1997). Frontal lobe volume loss observed with magnetic resonance imaging in older chronic alcoholics. Alcohol Clin Exp Res 21: 521–529.
Pfefferbaum A, Sullivan EV, Mathalon DH, Shear PK, Rosenbloom MJ, Lim KO (1995). Longitudinal changes in magnetic resonance imaging brain volumes in abstinent and relapsed alcoholics. Alcohol Clin Exp Res 19: 1177–1191.
Pfefferbaum A, Sullivan EV, Rosenbloom MJ, Mathalon DH, Lim KO (1998). A controlled study of cortical gray matter and ventricular changes in alcoholic men over a five year interval. Arch Gen Psychiatry 55: 905–912.
Pitkin SR, Savage LM (2001). Aging potentiates the acute and chronic neurological symptoms of pyrithiamine-induced thiamine deficiency in the rodent. Behav Brain Res 119: 167–177.
Plaitakis A, Nicklas WJ, Berl S (1978). Thiamine deficiency: selective impairment of the cerebellar serotonergic system. Neurology 28: 691–698.
Riley JN, Walker DW (1978). Morphological alterations in hippocampus after long-term alcohol consumption in mice. Science 201: 646–648.
Rintala J, Jaatinen P, Lu W, Sarviharju M, Eriksson CJ, Laippala P et al (1997). Effects of lifelong ethanol consumption on cerebellar layer volumes in AA and ANA rats. Alcohol Clin Exp Res 21: 311–317.
Rose SE, Nixon PF, Zelaya FO, Wholohan BT, Zimitat C, Moxon LN et al (1993). Application of high field localised in vivo1H MRS to study biochemical changes in the thiamine deficient rat brain under glucose load. NMR Biomed 6: 324–328.
Savage LM, Candon PM, Hohmann HL (2000). Alcohol-induced brain pathology and behavioral dysfunction: using an animal model to examine sex differences. Alcohol Clin Exp Res 24: 465–475.
Schroth G, Naegele T, Klose U, Mann K, Petersen D (1988). Reversible brain shrinkage in abstinent alcoholics, measured by MRI. Neuroradiology 30: 385–389.
Schroth G, Wichmann W, Valavanis A (1991). Blood–brain-barrier disruption in acute Wernicke encephalopathy: MR findings. J Comput Assist Tomogr 15: 1059–1061.
Schweinsburg B, Taylor M, Videen J, Alhassoon O, Patterson T, Grant I (2000). Elevated myo-inositol in gray matter of recently detoxified but not long-term abstinent alcoholics: a preliminary MR spectroscopy study. Alcohol Clin Exp Res 24: 699–705.
Schweinsburg BC, Alhassoon OM, Taylor MJ, Gonzalez R, Videen JS, Brown GG et al (2003). Effects of alcoholism and gender on brain metabolism. Am J Psychiatry 160: 1180–1183.
Schweinsburg BC, Taylor MJ, Alhassoon OM, Videen JS, Brown GG, Patterson TL et al (2001). Chemical pathology in brain white matter of recently detoxified alcoholics: a 1H magnetic resonance spectroscopy investigation of alcohol-associated frontal lobe injury. Alcohol Clin Exp Res 25: 924–934.
Seitz D, Widmann U, Seeger U, Nagele T, Klose U, Mann K et al (1999). Localized proton magnetic resonance spectroscopy of the cerebellum in detoxifying alcoholics. Alcohol Clin Exp Res 23: 158–163.
Shear PK, Sullivan EV, Lane B, Pfefferbaum A (1996). Mammillary body and cerebellar shrinkage in chronic alcoholics with and without amnesia. Alcohol Clin Exp Res 20: 1489–1495.
Sullivan EV (2000). Human brain vulnerability to alcoholism: evidence from neuroimaging studies. In: Noronha A, Eckardt M, Warren K (eds). Review of NIAAA's Neuroscience and Behavioral Research Portfolio NIAAA Research Monograph No. 34. National Institutes of Health: Bethesda, MD. pp 473–508.
Sullivan EV, Adalsteinsson E, Sood R, Mayer D, Bell RL, McBride WJ et al (2006). Part I: longitudinal brain MRI brain study of the alcohol-preferring (P) rat: adult brain growth. Alcohol Clin Exp Res (in press).
Sullivan EV, Deshmukh A, Desmond JE, Lim KO, Pfefferbaum A (2000a). Cerebellar volume decline in normal aging, alcoholism, and Korsakoff's syndrome: relation to ataxia. Neuropsychology 14: 341–352.
Sullivan EV, Deshmukh A, Desmond JE, Mathalon DH, Rosenbloom MJ, Lim KO et al (2000b). Contribution of alcohol abuse to cerebellar volume deficits in men with schizophrenia. Arch Gen Psychiatry 57: 894–902.
Sullivan EV, Lane B, Rosenbloom MJ, Deshmukh A, Desmond J, Lim KO et al (1999). In vivo mammillary body volume deficits in amnesic and nonamnesic alcoholics. Alcohol Clin Exp Res 23: 1629–1636.
Sullivan EV, Marsh L (2003). Hippocampal volume deficits in alcoholic Korsakoff's syndrome. Neurology 61: 1716–1719.
Sullivan EV, Marsh L, Mathalon DH, Lim KO, Pfefferbaum A (1995). Anterior hippocampal volume deficits in nonamnesic, aging chronic alcoholics. Alcohol Clin Exp Res 19: 110–122.
Sullivan EV, Rosenbloom MJ, Lim KO, Pfefferbaum A (2000c). Longitudinal changes in cognition, gait, and balance in abstinent and relapsed alcoholic men: relationships to changes in brain structure. Neuropsychology 14: 178–188.
Sullivan EV, Rosenbloom MJ, Serventi KL, Deshmukh A, Pfefferbaum A (2003). The effects of alcohol dependence comorbidity and anti-psychotic medication on volumes of the thalamus and pons in schizophrenia. Am J Psychiatry 160: 1110–1116.
Talland GA (1965). Deranged Memory; A Psychonomic Study of the Amnesic Syndrome. Academic Press: New York.
Thomson AD, Jeyasingham MD, Pratt OE, Shaw GK (1987). Nutrition and alcoholic encephalopathies. Acta Med Scand Suppl 717: 55–65.
Todd K, Butterworth RF (1999). Mechanisms of selective neuronal cell death due to thiamine deficiency. Ann NY Acad Sci 893: 404–411.
Torvik A, Lindboe CF, Rodge S (1982). Brain lesions in alcoholics: a neuropathological study with clinical correlations. J Neurol Sci 56: 233–248.
Torvik A, Torp S, Lindboe CF (1986). Atrophy of the cerebellar vermis in aging: a morphometric and histologic study. J Neurol Sci 76: 283–294.
Unlu E, Cakir B, Asil T (2006). MRI findings of Wernicke encephalopathy revisited due to hunger strike. Eur J Radiol 57: 43–53.
Victor M, Adams RD, Collins GH (1971). The Wernicke–Korsakoff Syndrome. Davis Co.: Philadelphia, PA.
Victor M, Adams RD, Collins GH (1989). The Wernicke–Korsakoff Syndrome and Related Neurologic Disorders Due to Alcoholism and Malnutrition, 2nd edn. Davis Co.: Philadelphia, PA.
Victor M, Herman K, White EE (1959). A psychological study of the Wernicke–Korsakoff syndrome. Results of Wechsler–Bellevue intelligence scale and Wechsler memory scale testing at different stages in the disease. Quart J Stud Alcohol 20: 467–479.
Webb P, Sailasuta N, Kohler S, Raidy T, Moats R, Hurd R (1994). Automated single-voxel proton MRS: technical development and multisite verification. Magn Reson Med 31: 365–373.
Wiggins RC, Gorman A, Rolsten C, Samorajski T, Ballinger WE, Freund G (1988). Effects of aging and alcohol on the biochemical composition of histologically normal human brain. Metab Brain Dis 3: 67–80.
Witt E (1985). Neuroanatomical consequences of thiamine deficiency: a comparative analysis. Alcohol Alcohol 20: 201–221.
Zelaya FO, Rose SE, Nixon PF, Wholohan BT, Bower AJ, Zimitat C et al (1995). MRI demonstration of impairment of the blood–CSF barrier by glucose administration to the thiamine-deficient rat brain. Magn Reson Imaging 13: 555–561.
Zhong C, Jin L, Fei G (2005). MR imaging of nonalcoholic Wernicke encephalopathy: a follow-up study. AJNR Am J Neuroradiol 26: 2301–2305.
Zhou FC, Lumeng L, Li TK (1995). Quantitative immunocytochemical evaluation of serotonergic innervation in alcoholic rat brain. Neurochem Int 26: 135–143.
Zimatkin SM, Zimatkina TI (1996). Thiamine deficiency as predisposition to, and consequence of, increased alcohol consumption. Alcohol Alcohol 31: 421–427.
Zimitat C, Kril J, Harper CG, Nixon PF (1990). Progression of neurological disease in thiamine-deficient rats is enhanced by ethanol. Alcohol 7: 493–501.
Acknowledgements
We thank Diane Howard, Wendy Baumgardner, Juan Orduna, Luke Garcia, Andrea Spadoni, and Daniel J Pfefferbaum for the invaluable care and time they devoted to various aspects of animal care, transportation, and scanning. We also thank Lisa Savage, PhD for advice and guidance in conducting the pyrithiamine experiment and Dirk Mayer, PhD for helpful consultation on shimming for MRS studies. Support for this project was provided by the Integrative Neuroscience Initiative on Alcoholism (INIA) from the National Institute on Alcohol Abuse and Alcoholism (AA13521 and AA13522) and by NIAAA AA05965.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pfefferbaum, A., Adalsteinsson, E., Bell, R. et al. Development and Resolution of Brain Lesions Caused by Pyrithiamine- and Dietary-Induced Thiamine Deficiency and Alcohol Exposure in the Alcohol-Preferring Rat: A Longitudinal Magnetic Resonance Imaging and Spectroscopy Study. Neuropsychopharmacol 32, 1159–1177 (2007). https://doi.org/10.1038/sj.npp.1301107
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301107
Keywords
This article is cited by
-
Recent Investigations on Neurotransmitters’ Role in Acute White Matter Injury of Perinatal Glia and Pharmacotherapies—Glia Dynamics in Stem Cell Therapy
Molecular Neurobiology (2022)
-
Comprehensive review of Wernicke encephalopathy: pathophysiology, clinical symptoms and imaging findings
Japanese Journal of Radiology (2020)
-
Concomitants of alcoholism: differential effects of thiamine deficiency, liver damage, and food deprivation on the rat brain in vivo
Psychopharmacology (2016)
-
Rat strain differences in brain structure and neurochemistry in response to binge alcohol
Psychopharmacology (2014)
-
Neuroanatomy and Neuropathology Associated with Korsakoff’s Syndrome
Neuropsychology Review (2012)
