
Abstract
The spatial genetic structure of a plant population provides a potential record of past gene flow and mating. We used hierarchical F-statistics and spatial autocorrelation to characterize spatial genetic differentiation of allozymes in adult Delphinium nuttallianum plants within and among six natural populations separated from one another by up to 3 km. Previous direct estimates suggested that gene flow is highly localized, averaging << 10 m. Earlier studies of seed-set, pollen-tube growth and progeny fitness suggested that partial reproductive isolation exists between plants growing too close together (<3 m) and too far apart (>100 m). Thus we anticipated substantial genetic differentiation on scales of a few to hundreds of metres. However, we detected little differentiation among the six populations, among replicate study plots within populations, or among subsections of study plots, except at the smallest scale of cm to m. These results suggest that relatively rare long-distance pollen movement has gone undetected and that postpollination selection may further modify genetic structure during the life cycle. Lack of differentiation is not at odds with the observation of partial reproductive isolation, because some loci may respond to spatial variation in selection without this response being evident at marker loci.
Similar content being viewed by others

Introduction
Patterns of mating and seed dispersal determine the initial spatial distribution of genotypes within and among plant populations (Wright, 1946, 1951; Crawford, 1984; Waller, 1993). It follows that spatial genetic structure may provide clues to past events of dispersal and mating, and this logic has engendered two well-recognized indirect methods for inferring gene dispersal, F-statistics (Wright, 1951; Weir & Cockerham, 1984) and spatial autocorrelation (Sokal & Oden, 1978). Although patterns of pollen and seed dispersal are broadly correlated with plant genetic structure (Hamrick & Godt, 1990), direct and indirect estimates of gene flow often disagree (e.g. Levin, 1981; Heywood, 1991; Slatkin, 1994; Williams, 1994; Berg & Hamrick, 1995). Such deviations from expectations based on the dispersal of neutral alleles provide insight into the myriad of forces that can influence genetic structure, including selection, genetic drift, inbreeding depression, and other postpollination events; furthermore they suggest fruitful avenues for additional research.
The larkspur Delphinium nuttallianum provides an example of the disagreement between direct and indirect estimates of gene flow. Pollinators appear to move most pollen << 10 m within populations of this species, leading to a prediction of genetic differentiation over distances of a few m to hundreds of m (Waser, 1987, 1988). Furthermore, reproductive success of plants, and long-term fitness of resulting offspring, are greatest following crosses over an intermediate distance, and decline both in crosses between neighbours and between plants tens to hundreds of m apart (Price & Waser, 1979; Waser & Price, 1991, 1994). Such spatially structured partial crossing barriers (or inbreeding and ‘outbreeding’ depression) independently suggest local genetic differentiation. However, an initial survey of allozymes detected little spatial genetic structure over distances up to ≈200 m (Waser, 1987).
In this paper we report on a more thorough study of genetic differentiation within and among six natural populations separated by 750–3250 m. We conclude, as did Waser (1987) that differentiation is minimal. We discuss some possible reasons for the discrepancy between direct and indirect estimates of gene flow, and point out why the lack of genetic structure does not necessarily contradict observations of inbreeding and outbreeding depression.
Methods
Natural history
Nuttall’s larkspur, Delphinium nuttallianum Pritzel (Ranunculaceae) (=D. nelsonii Greene), is a small herbaceous perennial common in dry subalpine meadows in the western USA. Plants flower from mid-May to early July near the Rocky Mountain Biological Laboratory (RMBL) in Colorado, at c. 2900 m elevation, typically producing 2–15 blue, zygomorphic, protandrous flowers in a single raceme. Plants do not reproduce vegetatively and there is virtually no dormant soil seed bank. Reproductive maturity is reached in 3–7 years or more (Waser & Price, 1994). The principal pollinators are broad-tailed hummingbirds (Selasphorus platycercus) and queen bumblebees (primarily Bombus appositus and B. flavifrons).
Population sampling
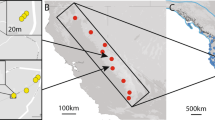
We sampled two populations in 1991 and 1992 and four more in 1995. Five of the six populations lie in the same East River Valley and span an approximate north–south transect 3250 m long (Fig. 1). The sixth population lies 1000 m up the tributary Copper Creek Valley. In each population we sampled two (in one case three) plots ≈100 m apart. Each plot was further subdivided into four subplots. With this design we were able to examine genetic differentiation on scales ranging from c. 1000 m (among populations) to ≈100 m (among replicate plots within populations) to ≈10 m (among subplots within plots) to << 10 m (individuals within mapped plots, see below).

Locations of the six Delphinium nuttallianum populations sampled in 1991–95 near the Rocky Mountain Biological Laboratory (RMBL). Population abbreviations are as follows: 401, Trail 401; CR, Carpenter Road; JF, Judd Falls; CC, Copper Creek; RM, Research Meadow; KP, Kettle Pond. Two plots (or three at KP) ≈100 m apart were sampled in each population. Expanded diagrams in the lower right-hand corner show the arrangement of 5 × 5 m quadrats in each of the two 20 × 40 m plots in the Judd Falls and Kettle Ponds populations sampled intensively in 1991 (JF A and B, KP A and B). The shaded central four 5 × 5 m quadrats in each plot were sampled most intensively.
The details are as follows. In 1991 we sampled two populations separated by 1500 m, hereafter Judd Falls (JF) and Kettle Pond (KP). In each population, two 20 × 40 m plots separated by 100 m (hereafter JF A and B; KP A and B) were gridded into 32 quadrats each of 5 × 5 m (Fig. 1 inset). We intensively sampled the central 10 × 10 m area of each plot, comprising four 5 × 5 m quadrats, by collecting leaf material from 165 to 200 flowering adults whose locations were mapped (two individuals m–2when present). When present, five adults were also sampled and mapped within each of the 28 surrounding quadrats (Fig. 1 inset, unshaded quadrats). Densities of adults were 7.7 m–2 in JF A, 11.7 m–2 in JF B, 8.2 m–2 in KP A, and 8.5 m–2 in KP B, and total samples ranged from 297 to 340 individuals per plot. In 1992 we sampled 161 adults in an additional plot (KP C) 200 m from the 1991 KP plots. In 1995 we sampled two plots 100 m apart in the Trail 401 (401 A and B), Carpenter Road (CR A and B), Copper Creek (CC A and B), and Research Meadow (RM A and B) populations. Each 1995 plot consisted of a 20-m transect along which we sampled 40 adults. We analysed data from all six populations to examine large-scale patterns of genetic differentiation, whereas data from the four intensively sampled 1991 plots alone were used for analysis of fine-scale genetic structure.
Electrophoresis
Leaf material was either preserved by freezing and lyophilizing for later processing, or kept on ice and processed within 48 h. We ground samples in ≈0.25 mL of buffer solution, soaked them into filter paper wicks and loaded them into gels. The grinding buffer contained 0.025 M L-ascorbic acid, 0.010 M MgCl2, 0.010 M KCl, 0.001 M EDTA (tetrasodium salt), 0.005 M sodium bisulphite, 0.006 M diethyldithiocarbamate, 0.200 M sucrose, 0.020% w/v PVP-40T, and 0.004% v/v β-mercaptoethanol, dissolved in 0.10 M Tris-HCl buffer, pH 8.0. Gels were 10% hydrolysed potato starch prepared in a microwave oven (Marty et al., 1984). We used two gel buffer systems: ‘AC buffer’, with an electrode buffer of 0.04 M citric acid, anhydrous, adjusted to pH 6.7 with N-3 aminopropyl-morpholine, and a gel buffer of 1:20 dilution of electrode buffer (modified from Marty et al., 1984); and ‘S6 buffer’, with an electrode buffer of 0.10 M NaOH, 0.30 M boric acid, pH 8.6, and a gel buffer of 0.015 M Tris, 0.004 M citric acid, anhydrous, at pH 7.8 (Soltis et al., 1983). Staining schedules followed Marty et al. (1984). We consistently resolved four enzyme systems coding for five polymorphic loci. In AC gels we scored two dimeric isozymes of malate dehydrogenase (Mdh-1 and Mdh-2), one dimeric isozyme of phosphoglucoisomerase (Pgi-2), and one monomeric isozyme of phosphoglucomutase (Pgm-1). In S6 gels we scored phosphoglucomutase (Pgm-1), and monomeric triose-phosphate isomerase (Tpi-3). Mdh-1 had two allozymes, Mdh-2 had six allozymes, Pgi-2 had eight allozymes, Pgm-1 had three allozymes and Tpi-3 had three allozymes. Alleles at all loci segregated in expected Mendelian diploid fashion in progeny arrays. In no case were there more than three common alleles per locus; for analysis we pooled rare alleles with the least common of these ‘major’ alleles.
Analyses
F-statistics We calculated single-locus and multilocus estimates of F-statistics (Weir & Cockerham, 1984; Williams & Guries, 1994). Single-locus estimates were tested for deviation from Hardy–Weinberg expectations using the methods of Li & Horvitz (1953) for F^IT and F^IS and Workman & Niswander (1970) for Θ^. 95% confidence intervals for multilocus F-statistics were calculated as 1.96 times the standard errors obtained by jackknifing over loci. We carried out three different analyses. First, we estimated hierarchical F-statistics for all six populations. In this analysis each population contained two plots (three for KP), and each plot contained four subplots. The two 20 × 40 m plots within each 1991 population were divided into four 10 × 20 m subplots, each of which included one 5 × 5 m section of the central intensively sampled area (Fig. 1 inset, shaded area). The additional KP C plot was divided into four subplots based on natural patches of plants. The two plots (=transects) in each of the four 1995 populations were divided into four subplots at ≈5 m intervals. We estimated genetic variance among individuals in the total sample (FIT), fixation within subplots (FIS), and differentiation among subplots within plots (ΘSUB), among plots within populations (ΘPLOT), and among populations within the total sample (ΘPOP). Secondly, we estimated these same hierarchical F-statistics for the 1991 JF and KP populations alone, to confirm that these intensively sampled populations yielded a pattern similar to that for all populations. Thirdly, to examine finer-scale patterns we estimated hierarchical F-statistics separately within each of the four 1991 plots. We divided each 20 × 40 m plot into eight 10 × 10 m subplots (Fig. 1, inset). We estimated differentiation among 10 × 10 m small subplots nested within 10 × 20 m medium subplots (ΘSMALL), among 10 × 20 m medium subplots within 20 × 20 m large subplots (ΘMEDIUM) and between the 20 × 20 m large subplots within the total 20 × 40 m plot (ΘLARGE).
Genetic distances
To examine further the relationship between geographical distance and genetic differentiation we calculated Edwards’s (1971) pairwise genetic distances between plots for all polymorphic loci and performed cluster analysis on genetic distances using the UPGMA method in the BIOSYS-1 computer program (Swofford & Selander, 1981).
Spatial autocorrelation
We used spatial autocorrelation (Sokal & Oden, 1978) to examine genetic variation at the smallest scales (a few cm to m). Spatial autocorrelations were calculated between individually mapped plants in each of the four 1991 plots using a program provided by J. S. Heywood. For each locus we calculated the autocorrelation coefficient, Moran’s I, for nearest neighbour pairs of individuals and all pairs of individuals in 1 m distance intervals. For diallelic loci, autocorrelations were calculated for only the most common allele. For multiallelic loci, autocorrelations were calculated for each allele separately. Alleles present at frequencies less than 0.01 were lumped with the least common allele present at a frequency ≥0.01. We constructed consensus correlograms for each plot by averaging the single-locus estimates in 1 m distance classes over the interval 0–20 m. To maintain statistical independence when averaging single-allele estimates, we dropped the least common allele at each locus. The null hypothesis that Moran’s I did not differ from the mean correlation among all individuals in the population was tested using the standard normal deviate (SND=[I – μ]/σ), under the assumption that samples are drawn at random from a normal distribution (Sokal & Oden, 1978). Single-allele values of |SND| ≥ 1.96 indicate that I differs significantly from the random expectation at P ≤ 0.05 (two-tailed).
Results
Large-scale differentiation within and among populations
Hierarchical F-statistics Single- and multilocus estimates were of small magnitude, with few single-locus estimates of Θ significantly different from zero. Single-locus estimates were generally concordant, and where they were not (Θ^SUB in all populations, Θ^PLOT and Θ^POP in 1991 populations), then multilocus estimates were not significant (Table 1). Genetic differentiation did not increase with greater geographical distance among subdivisions. Differentiation among subplots (Θ^SUB) 5–20 m apart was not significantly different from that among populations (Θ^POP) several km apart. Analyses of 1991 populations alone agreed with that of all six populations; in both cases overall genetic variance among individuals (F^IT) was mostly determined by fixation within subpopulations (F^IS), i.e. on a spatial scale of <10 m (Table 1).
Genetic distances
Pairwise Edwards’s genetic distances between the 13 plots ranged from ≈0.05–0.15, once again indicating only modest genetic differentiation. In three cases (KP, JF and 401), plots within the same population clustered together, but in the other three cases clustering was between plots from nonadjacent populations (not shown). Thus a strong and consistent relationship between genetic distance and physical separation was not apparent.
Small-scale differentiation within plots
Hierarchical F-statistics within 20 × 40 m plots The four 1991 JF and KP plots showed differentiation within plots and subplots similar to that at larger spatial scales, i.e. F^IS was the major contributor to F^IT in each plot (Table 2). Concordance among single-locus F-statistics within each plot was not as consistent as in the larger-scale analyses and few single-locus estimates were significant (Tables 1 and 2. Multilocus estimates of genetic differentiation at all hierarchical levels were small, and significant only at small or medium spatial scales (Table 2). In all but KP B the greatest differentiation occurred among the smallest subplots. In KP B more genetic variance occurred between the largest subplots, but this estimate was not significantly different from zero. Once again, therefore, most genetic differentiation appeared to be organized at a scale <10 × 10 m. We also detected differences in structure among plots (Table 2). The two KP plots had fixation indices within 10 × 10 m subplots approximately twice as large as those in the JF plots, and somewhat higher levels of differentiation among small subplots than the JF plots.
Spatial autocorrelation within 20 × 40 m plots
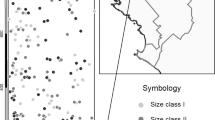
Of 37 nearest-neighbour autocorrelation coefficients, Moran’s INN, from individual alleles in the 1991 JF and KP plots, 23 were positive and 14 negative (Table 2). Nine positive values were individually significant at P < 0.05. Several significant positive values belonged to rare alleles, suggesting that such alleles tend to be clumped at this scale. The significant positive autocorrelations were concentrated in the KP B plot. Whereas the mean values of INN across alleles were very small or negative for both JF plots and for KP A, the mean for KP B was 0.155, indicating the strongest local kinship structure in that plot (Table 2). A similar story is told by consensus correlograms for distance classes out to 20 m (Fig. 2). The shortest distance class in correlograms includes all joins between plants separated by up to 1 m, including nearest neighbours. On average, values of I were weakly positive for this distance class in JF A and KP A plots and weakly negative in JF B. In contrast, I at the shortest distance class in KP B was positive and approximately five times as large as any of the other values. Average values of I remained positive at KP B to the third distance class (2.01–3 m), unlike the case at other sites. Finally, the numbers of single-allele estimates that differed significantly from random in each distance class also varied among plots (Table 3). Most of the significant positive single-allele estimates were in the KP plots in the first 5–6 m. Few significant values were found beyond 10 m, and hence Table 3 omits these longer distances. The KP B plot had six of seven alleles with significant I in the 0–1 m distance class. This again suggests that KP B had more local genetic structure than the other plots.

Consensus correlograms of Moran’s I in 1 m distance intervals averaged over alleles at the four plots of Delphinium nuttallianum sampled in 1991. The least common allele at each locus was dropped before computing averages. Significance tests of single- allele estimates used to calculate averages are in Table 3.
Discussion
We anticipated substantial genetic differentiation within and among populations of D. nuttallianum, because direct estimates indicated that gene dispersal is limited. Seed dispersal is predominantly passive, averaging ≈10 cm (Waser & Price, 1983). Median distances flown by hummingbird and bumble bee pollinators between successive flowers are ≤1 m, matching median dispersal of fluorescent dye particles, whereas pollen is transported 1.5–1.8 times further than dye (Waser, 1988). From this information, and applying a correction for inbreeding and outbreeding depression in seed-set, Waser (1988) arrived at ≈ 85 plants in an area of 17 m2 for the genetic neighbourhood. With such small neighbourhoods one expects isolation by distance within continuous populations for neutral alleles (Wright, 1946; Crawford, 1984; Heywood, 1991), whereas at larger spatial scales random genetic drift and/or spatially heterogeneous selection should produce variation among population subdivisions (Wright, 1951; Cockerham & Weir, 1993; Slatkin, 1994). However, Waser (1987) reported only minor genetic differentiation among putative genetic neighbourhoods. The more thorough investigation described here reaches the same conclusion. We detected only small amounts of genetic differentiation within and among populations. Our estimates of ≈0.005–0.05 for F-statistics, and ≤0.1 for Moran’s I between near-neighbours are at the low end of the range of estimates reported for widely dispersed species with predominantly outcrossing mating systems (Hamrick & Godt, 1990; Heywood, 1991; Williams, 1994). Furthermore, genetic differentiation was not consistently related to physical separation, as predicted by isolation-by-distance or stepping-stone models of population structure (Wright, 1946; Kimura & Weiss, 1964). How can we explain this discrepancy between direct estimates of restricted gene flow, and lack of strong detectable genetic structure?
One simple possibility is that actual gene flow exceeds our direct estimates, as do actual neighbourhood sizes. This possibility is in accord with the results from other outcrossing species, in which gene flow appears sufficient to limit the isolation-by-distance process severely (Heywood, 1991; Williams, 1994; Berg & Hamrick, 1995). We suspect that our earlier studies of D. nuttallianum failed to detect relatively rare long-distance pollinator flights (Levin, 1981; Ellstrand et al., 1989; Broyles et al., 1994). An experiment by Schulke (1999) hints at the possible effect of such flights on neighbourhood size within continuous populations, and on the likelihood of movement among disjunct populations such as those we sampled at the largest spatial scale. Schulke recorded high and consistent levels of pollen receipt and seed-set in arrays of potted D. nuttallianum plants with emasculated flowers that were placed up to 400 m from the nearest natural population. He also found that ≈5% of all pollinator flights moved beyond the range of an observer, and showed how estimates of genetic neighbourhood size would rapidly rise into the thousands if some of these flights were as long as several hundred m.
Despite meagre genetic differentiation at larger spatial scales, we did find significant genetic structure at the smallest scales of cm to m, although the pattern varied among plots and to a lesser degree among loci. We interpret this as a result of kinship in the seed crops of individual plants. Progeny within seed crops are related minimally as maternal half-sibs. Because most seeds germinate directly under the maternal parent (Waser & Price, 1983), we anticipate transient local kin structure that is regenerated during every episode of seedling recruitment. Such structure is predicted even if gene dispersal through pollen is substantial (Van Dijk, 1987), and has been demonstrated for maternally inherited genes in other species (e.g. McCauley, 1997; Latta et al., 1998). The variation in fine-scale genetic structure among our replicate plots may also help explain differences in the magnitude of inbreeding depression estimated in hand-pollination experiments conducted with D. nuttallianum at different times and localities (Price & Waser, 1979; Waser & Price, 1983, 1991, 1994).
It is also important to recognize that processes occurring after pollination can modify the patterns established by patterns of mating and gene flow. For example, the success of pollen tubes in growing to the ovary and the final production of seeds both vary with the distance between the two parental D. nuttallianum plants (Waser & Price, 1991). The direct estimate of neighbourhood size described above does incorporate these effects, but not the possible effect of mixing pollen from several donors on the same stigma, as often occurs in nature (e.g. Rigney, 1995; Mitchell & Marshall, 1998). Pollen arriving from longer distances might gain a stronger advantage in mixtures, thus elevating effective pollen dispersal (Rigney, 1995). Postfertilization selection might further modify the final realized genetic structure measured in adults (Clegg et al., 1978). Such selection might arise from inbreeding depression (and heterosis) or outbreeding depression, with genome-wide effects on genotypic arrays, or it might act on marker alleles directly. In the latter regard, our single-locus results do not indicate any strong selection on marker loci: although we detected heterogeneity among single-locus estimates, no locus was consistently different from the others in its pattern of variation across replicate plots, as expected with single-locus selection. Whatever the mechanism, selection between seed maturation and sexual maturity is expected to alter the genetic structure present in the seed crop (Eguiarte et al., 1992; Tonsor et al., 1993; Stanton et al., 1997). Alternatively, genetic structure might decay over the life cycle because of sampling artifacts (Alvarez-Buylla et al., 1996). In the future we intend to explore these possibilities using a genetic demography approach.
Finally, it may appear that there is another discrepancy to explain in the D. nuttallianum system: that between the existence of outbreeding depression and the lack of strong spatial genetic structure among populations. However, a lack of detectable structure at neutral or nearly neutral markers, as we have argued our electrophoretic markers to be, in no way implies lack of structure at all loci. Local adaptation may evolve over a wide range of spatial scales (reviewed in Stanton & Galen, 1997). Loci conferring adaptation to local edaphic and biotic conditions, for example, may show strong spatial differentiation despite little genetic structure at neutral loci, because the amount of gene flow necessary to counteract directional selection is far greater than that needed to counteract random genetic drift (Wright, 1932; May et al., 1975). Neutral markers can hitchhike on selected loci, but only with tight physical linkage or large selection coefficients (Clegg, 1984), and so are unlikely to mirror structure at loci under spatially varying selection (but see Tonsor et al., 1993). In fact, a reciprocal seed transplantation experiment with D. nuttallianum did demonstrate substantial local adaptation, and by inference genetic differentiation, over a scale of 50 m (Waser & Price, 1985). Disruption of such local adaptation offers an explanation for our observations of outbreeding depression in this species, in spite of near-panmixia at marker loci.
References
Alvarez-Buylla, E. R., Chaos, A., Piñero, D. and Garay, A. A. (1996). Demographic genetics of a pioneer tropical tree species: patch dynamics, seed dispersal, and seed banks. Evolution. 50: 1155–1166.
Berg, E. E. and Hamrick, J. L. (1995). Fine-scale genetic structure of a turkey oak forest. Evolution. 49: 110–120.
Broyles, S. B., Schnabel, A. and Wyatt, R. (1994). Effective pollen dispersal in a natural population of Asclepias exaltata: the influence of pollinator behavior, genetic similarity, and mating success. Am Nat. 138: 1239–1249.
Clegg, M. T. (1984). Dynamics of multilocus genetic systems. Oxford Surveys in Evolutionary Biology. 1: 160–183.
Clegg, M. T., Kahler, A. L. and Allard, R. W. (1978). Genetic demography of plant populations. In: Brussard, P. F. (ed.) Ecological Genetics: The Interface pp. 173–188. Springer, New York.
Cockerham, C. C. and Weir, B. S. (1993). Estimation of gene flow from F-statistics. Evolution. 47: 855–863.
Crawford, T. J. (1984). What is a population?. In: Shorrocks, B. (ed.) Evolutionary Ecology pp. 135–173. Blackwell Scientific Publications, Oxford.
Edwards, A. W. F. (1971). Distances between populations on the basis of gene frequencies. Biometrics. 27: 873–881.
Eguiarte, L. E., Perez-Nassar, N. and Piñero, D. (1992). Genetic structure, outcrossing rate and heterosis in Astrocaryum mexicanum (tropical palm): implications for evolution and conservation. Heredity. 69: 217–228.
Ellstrand, N. C., Devlin, B. and Marshall, D. L. (1989). Gene flow by pollen into small populations: data from experimental and natural stands of wild radish. Proc Natl Acad Sci USA. 86: 9044–9047.
Hamrick, J. L. and Godt, M. J. W. (1990). Allozyme diversity in plant species. In: Brown, A. H. D., Clegg, M. T., Kahler, A. L. and Weir, B. S. (eds) Population Genetics, Breeding, and Genetic Resources pp. 43–63. Sinauer Associates, Sunderland, MA.
Heywood, J. S. (1991). Spatial analysis of genetic variation in plant populations. Ann Rev Ecol Syst. 22: 335–355.
Kimura, M. and Weiss, G. H. (1964). The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics. 49: 561–576.
Latta, R. G., Linhart, Y. B., Fleck, D. and Elliot, M. (1998). Direct and indirect estimates of seed versus pollen movement within a population of ponderosa pine. Evolution. 52: 61–67.
Levin, D. A. (1981). Dispersal versus gene flow in plants. Ann Mo Bot Gard. 68: 233–253.
Li, C. C. and Horvitz, D. G. (1953). Some methods of estimating the inbreeding coefficient. Am J Hum Genet. 5: 107–117.
Marty, T. L., O’malley, D. M. and Guries, R. P. (1984). A Manual for Starch-Gel Electrophoresis: New Microwave Edition University of Wisconsin Department of Forestry Staff Paper Series, no. 20.
May, R. M., Endler, J. A. and Mcmurtrie, R. E. (1975). Gene frequency clines in the presence of selection opposed by gene flow. Am Nat. 109: 659–676.
McCauley, D. E. (1997). The relative contributions of seed and pollen movement to the local genetic structure of Silene alba. J Hered. 88: 257–263.
Mitchell, R. J. and Marshall, D. L. (1998). Nonrandom mating and sexual selection in a desert mustard: an experimental approach. Am J Bot. 85: 48–55.
Price, M. V. and Waser, N. M. (1979). Pollen dispersal and optimal outcrossing in Delphinium nelsoni. Nature. 277: 294–297.
Rigney, L. P. (1995). Postfertilization causes of differential success of pollen donors in Erythronium grandiflorum (Liliaceae): nonrandom ovule abortion. Am J Bot. 82: 578–584.
Schulke, B. (1999). Experimental Studies of Long-Distance Pollen-Mediated Gene Flow in Delphinium nelsonii. M.Sc. Thesis, University of California, Riverside.
Slatkin, M. (1994). Gene flow and population structure. In: Real, L. A. (ed.) Ecological Genetics pp. 3–17. Princeton University Press, Princeton, NJ.
Sokal, R. R. and Oden, N. L. (1978). Spatial autocorrelation methods in biology. I. Methodology. Biol J Linn Soc. 10: 199–228.
Soltis, D. E., Haufler, C. H., Darrow, D. C. and Gastony, G. J. (1983). Starch gel electrophoresis of ferns: a compilation of grinding buffers, gel and electrode buffers, and staining schedules. Am Fern J. 73: 9–27.
Stanton, M. L. and Galen, C. (1997). Life on the edge: adaptation versus environmentally mediated gene flow in the Snow Buttercup, Ranunculus adoneus. Am Nat. 150: 143–178.
Stanton, M. L., Galen, C. and Shore, J. (1997). Population structure along a steep environmental gradient: consequences of flowering time and habitat variation in the Snow Buttercup, Ranunculus adoneus. Evolution. 51: 79–94.
Swofford, D. L. and Selander, R. B. (1981). BIOSYS-1: a computer program for the analysis of allelic variation in population genetics and biochemical systematics Release 1.7. Illinois Natural History Survey, Champaign, IL.
Tonsor, S. J., Kalisz, S., Fisher, J. and Holtsford, T. P. (1993). A life-history based study of population genetic structure: seed bank to adults in Plantago lanceolata. Evolution. 47: 833–843.
van Dijk, H. (1987). A method for the estimation of gene flow parameters from a population structure caused by restricted gene flow and genetic drift. Theor Appl Genet. 73: 724–736.
Waller, D. M. (1993). The statics and dynamics of mating system evolution. In: Thornhill, N. W. (ed.) The Natural History of Inbreeding and Outbreeding: Theoretical and Empirical Perspectives pp. 97–117. University of Chicago Press, Chicago, IL.
Waser, N. M. (1987). Spatial genetic heterogeneity in a population of the montane perennial plant Delphinium nelsonii. Heredity. 58: 249–256.
Waser, N. M. (1988). Comparative pollen and dye transfer by natural pollinators of Delphinium nelsonii. Funct Ecol. 2: 41–48.
Waser, N. M. and Price, M. V. (1983). Optimal and actual outcrossing in plants, and the nature of plant–pollinator interaction. In: Jones, C. E. and Little, R. J. (eds) Handbook of Experimental Pollination Biology pp. 341–359. Van Nostrand Reinhold, New York.
Waser, N. M. and Price, M. V. (1985). Reciprocal transplant experiments with Delphinium nelsonii (Ranunculaceae): evidence for local adaptation. Am J Bot. 72: 1726–1732.
Waser, N. M. and Price, M. V. (1991). Outcrossing distance effects in Delphinium nelsonii: pollen load, pollen tubes, and seed set. Ecology. 72: 171–179.
Waser, N. M. and Price, M. V. (1994). Crossing distance effects in Delphinium nelsonii: outbreeding and inbreeding depression in progeny fitness. Evolution. 48: 842–852.
Weir, B. S. and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution. 38: 1358–1370.
Williams, C. F. (1994). Genetic consequences of seed dispersal in three sympatric forest herbs. II. Microspatial genetic structure within populations. Evolution. 48: 1959–1972.
Williams, C. F. and Guries, R. P. (1994). Genetic consequences of seed dispersal in three sympatric forest herbs. I. Hierarchical population-genetic structure. Evolution. 48: 791–805.
Workman, P. L. and Niswander, J. D. (1970). Population studies on southwestern Indian tribes. II. Local genetic differentiation in the Papago. Am J Hum Genet. 22: 24–49.
Wright, S. (1932). The roles of mutation, inbreeding, crossbreeding and selection in evolution. Proc Sixth Int Cong Genet. 1: 356–366.
Wright, S. (1946). Isolation by distance under diverse systems of mating. Genetics. 31: 39–59.
Wright, S. (1951). The genetical structure of populations. Ann Eugen. 15: 323–354.
Acknowledgements
For assistance in field and laboratory, discussions, and comments we thank S. Airamé, H. Callahan, T. Crawford, D. Janus, J. Joyner, R. Mitchell, M. Price, H. Renkin, B. Schulke, R. Smith, S. Szychowski, and J. Ruvinsky and two anonymous reviewers. Support was provided by NSF grants BSR 8905808 to N.M.W. and BSR 9103781 to C.F.W., the Howard Hughes Medical Institute, Nebraska Wesleyan University, and the University of California, Riverside Academic Senate.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Williams, C., Waser, N. Spatial genetic structure of Delphinium nuttallianum populations: inferences about gene flow. Heredity 83, 541–550 (1999). https://doi.org/10.1038/sj.hdy.6885920
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6885920
