
Abstract
Large-scale evaluations of genetic diversity in domestic livestock populations are necessary so that region-specific conservation measures can be implemented. We performed the first such survey in European sheep by analysing 820 individuals from 29 geographically and phenotypically diverse breeds and a closely related wild species at 23 microsatellite loci. In contrast to most other domestic species, we found evidence of widespread heterozygote deficit within breeds, even after removing loci with potentially high frequency of null alleles. This is most likely due to subdivision among flocks (Wahlund effect) and use of a small number of rams for breeding. Levels of heterozygosity were slightly higher in southern than in northern breeds, consistent with declining diversity with distance from the Near Eastern centre of domestication. Our results highlight the importance of isolation in terms of both geography and management in augmenting genetic differentiation through genetic drift, with isolated northern European breeds showing the greatest divergence and hence being obvious targets for conservation. Finally, using a Bayesian cluster analysis, we uncovered evidence of admixture between breeds, which has important implications for breed management.
Similar content being viewed by others
Introduction
Sheep (Ovis aries, L.) were domesticated from at least three ancestral subspecies of the wild Mouflon (O. gmelini, Gmelin 1774) approximately 9000 years before present (YBP) in Southwest Asia, and by 5000 YBP, sheep had been transported throughout Europe (Ryder, 1981; Hiendleder et al., 1998, 2002; Guo et al., 2005; Pedrosa et al., 2005; Bruford and Townsend, 2006). Today, over 850 sheep breeds are recognized worldwide, and Europe supports a greater number of breeds than any other continent (United Nations Food and Agriculture Organization, FAO, 2000; Rege and Gibson, 2003).
Over the past few decades though, it has become apparent that many of these breeds are at risk of extinction. Europe faces the greatest threat, with 18% of its livestock breeds lost in the last century alone and a further 40% at risk of becoming extinct over the next 20 years (compared to 32% at risk worldwide, FAO, 2000; Signorello and Pappalardo, 2003). Intensive production and increased commercial demands, particularly since the end of the Second World War, have significantly contributed to the threats facing European sheep breeds. First, artificial insemination and improved transportation have reduced the number of breeding rams, leading to a reduction in the effective population size (Ne) of many breeds. Second, production has focused on only a few breeds, to the detriment of rare or minority breeds, which are likely to be important genetic resources because of their local adaptation, disease resistance, high fertility and unique product qualities (Mendelsohn, 2003). Minority breeds have also been lost by introgression into large commercial populations.
The loss of diversity in domestic species has important economic, ecological and scientific implications as well as social considerations, and in response to these threats, the FAO, through its Domestic Animal Diversity Information System (DAD-IS), initiated a programme to document the State of the World's Animal Genetic Resources (SoWAnGR, FAO, 2000). An understanding of the evolutionary history of domestic breeds and data on genetic variation within and among breeds is vital to these initiatives to provide critically important data for the decision-making process (Rege and Gibson, 2003). Information on both within- and among-breed diversity is important. The former provides information for management at the breed level. The latter helps to identify divergent breeds that may harbour distinct genotypes and are, therefore, worthy of conservation efforts even if their within-breed diversity is relatively high.
To date, most studies of genetic diversity in livestock species have been carried out at local geographic (national) scales (Arranz et al., 1998; Martín-Burriel et al., 1999; Saitbekova et al., 1999; Martinez et al., 2000; Mateus et al., 2004), or on a small number of breeds (MacHugh et al., 1998; Diez-Tascón et al., 2000; Pariset et al., 2003). While such studies are essential for region- or breed-specific management and conservation programmes, it is also important to assess how genetic diversity is partitioned at larger geographic scales to better implement region-specific conservation measures (Bruford et al., 2003).
In the present study, we use a panel of 23 autosomal microsatellite markers to evaluate how genetic diversity is partitioned within and among a diverse sample of 30 European sheep breeds. We consider the role of geography and breed type in determining diversity and differentiation, and examine the extent of admixture among breeds in relation to conservation and management.
Materials and methods
Sampling procedure and DNA extraction
A total of 820 individuals from 29 breeds of domestic sheep (O. aries) and one feral, putatively ancestral type, the European Mouflon (O. gmelini musimon), were analysed to ensure sampling of morphologically and geographically divergent types. Our aim was to collect at least 30 samples from a minimum of two separate flocks, although fulfilment of these objectives was not possible for all breeds (Table 1).
Samples (from blood, tissue and hair) and pre-extracted DNA were obtained from farmers and collaborators. Blood samples were diluted with five volumes of lysis buffer (Scott and Densmore, 1991) and DNA extracted following Sambrook et al. (1989). Tissue samples (such as heart and muscle collected as by-products from animals that had been slaughtered) were extracted using a sodium dodecyl sulphate–ethylenediaminetetraacetic acid (SDS–EDTA) procedure, as described by Hoelzel and Dover (1991). Hair samples (collected from live animals) were extracted using an SDS–EDTA procedure or a Chelex 100-based method (Bio-Rad, Walsh et al., 1991).
Microsatellite loci
Twenty-three microsatellite loci (Table 2) were chosen from the literature following three criteria: (1) a similar proportion of markers were derived from sheep, cattle and goat (to demonstrate cross-species utility of markers for the purposes of the European Union); (2) loci were distributed evenly throughout the autosomes and preferably unlinked; and (3) loci exhibited Mendelian inheritance and at least four alleles. For four of the breeds (Zeeland, Exmoor Horn, Llanwenog and Bizet), data were analysed for a subset of 10 of these loci (OarAE54, OarAE129, OarFCB20, OarFCB304, JMP29, JMP58, MAF65, MAF70, MAF209 and BM1824) that were typed in a collaborative study (K Byrne, unpublished results).
Polymerase chain reaction (PCR) amplifications were carried out in 10 μl total volumes, containing 2–4 pM of each primer (with the forward primer labelled with 5-FAM, TET or HEX), 0.275 mM dNTPs (Pharmacia), 1 × NH4 buffer, 2–4.0 mM MgCl2 (Table 2), 0.25 U of Amplitaq Gold DNA polymerase (Applied Biosystems, Foster City, CA, USA) and ∼50 ng DNA. PCRs were performed in a PE9700 thermocycler (Applied Biosystems), with the following cycling parameters: 94°C for 2 min followed by 30 cycles of 94°C for 30 s, anneal (Table 2) for 30 s, and 72°C for 30 s, and a final step at 72°C for 10 min. For some loci (identified by two annealing temperatures separated by ‘/’ in Table 2), a two-step cycling procedure was used, which consisted of 94°C for 6 min, followed by seven cycles of 94°C for 30 s, anneal (Table 2) for 30 s, and 72°C for 1 min, then 23 cycles of 92°C for 30 s, anneal (Table 2) for 30 s, and 72°C for 1 min, and final extension at 72°C for 10 min.
Following PCR, products were diluted 1/10–1/20 and 0.8 μl was mixed with an internal standard (TAMRA, Applied Biosystems) according to the manufacturer's instructions. Products were electrophoresed on a 4.25% polyacrylamide gel in an ABI377 automated sequencer (Applied Biosystems). Fragment analysis was carried out using GENESCAN v3.1 and GENOTYPER v2.0 software (Applied Biosystems). Genotyping was repeated once when individuals failed to amplify.
General statistical analyses
Allelic richness was estimated over all samples for each locus (Rt), without correction for null alleles, using the rarefaction technique of El Mousadik and Petit (1996) in FSTAT (v2.9.3; Goudet, 2000). Genotypic linkage disequilibrium (LD) was assessed using the exact test in GENEPOP (v3.4; Raymond and Rousset, 1995; Rousset and Raymond, 1995), using a Markov chain with 10 000 dememorization steps, 200 batches and 2000 iterations per batch to produce an unbiased estimate of the exact P-value.
Null alleles––alleles that consistently fail to amplify during PCR due, for example, to priming site mutations, differential amplification of size variants or inconsistent DNA quality––can decrease estimates of genetic diversity and inflate genetic differentiation, particularly when loci characterized in one species are then analysed in a different species, as is the case here (Chakraborty et al., 1992; Paetkau and Strobeck, 1995; Slatkin, 1995; Dakin and Avise, 2004). To investigate whether null alleles were present in our data set, we first recorded the number of times amplification failed for just one locus per individual, which could signify a null homozygote. We then estimated the potential frequency of null alleles (r) for each locus in each breed using the EM algorithm of Dempster et al. (1977) in the software FreeNA (Chapuis and Estoup, 2007). Note that this method, along with all other methods for detecting null alleles, assumes that deviations from Hardy–Weinberg (HW) equilibrium do not result from other causes (such as a Wahlund effect). Values of r⩽0.2 are not expected to cause significant problems in analyses (Dakin and Avise, 2004; Chapuis and Estoup, 2007); therefore, we only considered loci with r⩾0.2 to be potentially problematic for our calculations.
Genetic diversity within and among breeds
Observed and expected heterozygosities (Ho and He, respectively) were calculated for each breed and locus combination in ARLEQUIN (v2.0; Schneider et al., 2000) and then averaged over loci to obtain an estimate per breed. Deviation from HW equilibrium was assessed using the method of Guo and Thompson (1992) for each locus and breed combination in ARLEQUIN using a Markov chain of 100 000 steps and 1000 dememorization steps. GENEPOP was used to assess global deviations from HW equilibrium (over all loci per breed and after Bonferroni correction) using the same Markov chain parameters as for the genotypic disequilibrium test. Weir and Cockerham's (1984) estimate of Fis was calculated per locus and over all loci per breed using FSTAT and P-values obtained based on 2300 randomizations (number of loci, nl × 100). Ho, He and Fis were obtained, and deviations from HW equilibrium were assessed before and after removal of loci (on a per-breed basis) that showed potentially high null allele frequencies (r⩾0.2).
Since heterozygote deficit was widespread even after removing loci with r⩾0.2 (see Results and Table 4), and since excluding these loci greatly reduced the amount of available data for pairwise comparisons among breeds, all loci were retained for analyses described below. Pairwise genetic distance between breeds was estimated using Weir and Cockerham's (1984) estimate of Wright's Fst (θ, Wright, 1951, 1977) and P-values obtained using 435 000 permutations (number of tests × 100) and standard Bonferroni correction in FSTAT.
Genetic variation was compared among groups, among breeds within groups and within breeds using a hierarchical Analysis of MOlecular VAriance (AMOVA; Excoffier et al., 1992) in ARLEQUIN. Significance levels were determined after 1000 permutations. Breeds were partitioned in three ways: by main geographic region; by north versus south; and by breed type. The main geographic regions were (a) Near East (Israel and Jordan), (b) Greece and Cyprus, (c) Italy and Croatia, (d) Spain and France, (e) Romania, Hungary and Czech Republic, (f) Germany and The Netherlands and (g) The UK and Iceland (Table 1). Southern breeds comprised groups (a–e) from the main geographic regions above and northern breeds comprised groups (f–g) (Table 1). Breed types were fat-tail, Zackel, coarse wool, mountain, heath, marsh island rat-tail and northern short-tail (Table 1). Breeds of unique type or sole representatives of their type were excluded from the analysis (Table 1). Several other AMOVAs were performed using different regional group definitions (for example, mainland versus island breeds), but among group differences were not significant and they are, therefore, not presented.
Relationships among breeds and admixture
A principal component analysis (PCA) was performed to visualize pairwise differentiation among breeds (Fst) using the software PCAGEN (J Goudet, personal communication, available at http://www2.unil.ch/popgen/softwares/pcagen.htm). In total, 1000 randomizations of genotypes were performed to test for significance of axes.
Admixture was investigated using the Bayesian clustering algorithm implemented in STRUCTURE (Pritchard et al., 2000). The full data set of 30 breeds was run with K=1–30, where K is the potential number of genetic clusters. Breeds that shared the same genetic cluster were then analysed independently with K=1–6. A total of 100 000 iterations of the Gibbs sampler were run, after a burn-in of 50 000 iterations using the admixture option. The program DISTRUCT (Rosenberg, 2004) was used to graphically display the membership coefficient of an individual for a sub-population (i.e. breed), which represents the fraction of its genome that has ancestry in the sub-population, (Rosenberg, 2004).
Results
The number of alleles per locus ranged from 10 for BM1824 to 33 for MAF214 (Table 2). Loci derived from sheep had more alleles and higher overall allelic richness (Rt) than cattle or goat loci (mean number of alleles 22.45, 17.50 and 17.67 respectively, Rt 9.07, 6.02, 5.94). In the case of Rt, but not allele number, this difference was significant (Mann–Whitney U score test, sheep/cattle U=56, P=0.02, sheep/goat U=58, P=0.01). Significant LD was found in comparisons of OarAE129/ILSTS028, OarJMP58/INRA063, BM1824/SRCRSP3 and ILSTS005/SRCRSP7 and SRCRSP3, and ILSTS028/INRA063; however, in each case, the loci were situated on different chromosomes.
Twenty-five breeds had at least one individual that failed to amplify at only one locus, which could signify a null homozygote. Of the 15 loci that were affected, SRCRSP5 was the worst offender (being the sole non-amplifying locus in 25/702 individuals in which it was typed, which accounts for 3.6% of genotypes) followed by ILSTS11, ILSTS28, SRCRSP8 and OarFCB48 (accounting for 2.4, 2.4, 2.2 and 2.0% of genotypes, respectively; see Supplementary Table 1 for details). Using the EM method (Dempster et al., 1977; Chapuis and Estoup, 2007), we identified 12 loci that had potential null alleles at high frequency (r⩾0.2) in at least one breed (Supplementary Table 2, summarized in Table 3). OarAE54 had r⩾0.2 in four breeds (Lesvos, Argos, Comisana and Mouflon), OarAE129 and SRCRSP5 had r⩾0.2 in three breeds (Argos, Skudde, Heidschnuke and Comisana, Lecesse, Sarda, respectively), and the remaining nine loci had r⩾0.2 in one or two breeds only (Table 3). Note that OarAE129 and SRCRSP5 have been shown to produce null alleles in other studies (Peter et al., 2005, 2007).
Genetic diversity within and among breeds
After exclusion of the loci with r⩾0.2, observed heterozygosity ranged from 0.495 to 0.711 (mean Ho=0.620, s.d. 0.060; Table 3), with the lowest values found in the North Ronaldsay, Friesland and Soay, and the highest in the Istrian, Leccese and Tsigai. Breeds from southern Europe had significantly higher levels of heterozygosity (mean Ho=0.641, s.d. 0.044) than northern breeds (mean Ho=0.589, s.d. 0.073, Mann–Whitney U=50, P=0.04). Out of a possible 613 locus × breed combinations (again excluding the loci with r⩾0.2), heterozygote excess was found on only 16 occasions, compared to 199 instances of heterozygote deficit (Table 4 and Supplementary Table 3). On average, roughly nine loci per breed showed heterozygote deficit (from the total number of loci per breed with heterozygote deficit divided by the total number of locus × breed combinations, multiplied by the number of breeds, to the nearest integer: (183/613) × 30=9). This translates to significant global heterozygote deficit in all breeds (Table 3, unbiased P-values for tests of global heterozygote deficit are P<0.000 for all breeds except Soay and Churro, where P=0.002) except two with small sample size (n<15, Istrian and Heidschnuke, where P=0.541 and 0.449, respectively). With the exception of these two, most breeds retained positive, significant values of Fis after correction for null alleles (Table 3). Excluding loci with r⩾0.2 increases Ho and decreases values of Fis, but does not change the significance of any comparison (Table 3). Since it therefore seems unlikely that null alleles are the only cause for deviations from HW equilibrium, we based the following results on all loci.
Global Fst was 0.131 (95% confidence interval 0.116–0.147), while pairwise values ranged from 0.01 to 0.275 (Turcana/Tsigai and Soay/Friesland, respectively; Table 6). All comparisons except Turcana/Tsigai, Thônes-Marthod/Leccese and Thônes-Marthod/Istrian were significant after Bonferroni correction (P<0.05; Table 6). Northern European breeds such as the Soay, Friesland and Skudde tended to show the greatest divergence (Fst>0.20).
Relationships among breeds and admixture analysis
Our hierarchical AMOVA with hypothetical population structure revealed that the greatest variation among groups corresponds to breed type (2.7%, P<0.0001; Tables 5 and 6) as opposed to region (0.97%, P<0.05), or north versus south (1.16%, P<0.0001); however, most of the variation was consistently explained within breeds (∼87%, P<0.0001).
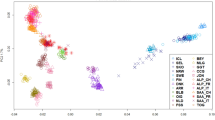
The PCA shown in Figure 1 illustrates that most breeds cluster within a central group, with little correspondence to breed type (except for Zackel breeds; Turcana, Tsigai, Racka and Šumavka). The most distant outliers in the PCA (Friesland, North Ronaldsay, Icelandic, Soay and Skudde) are all from northern Europe.

Principal component analysis (PCA) based on pairwise Fst. Both axes are significant. Corresponding Fst values are: axis 1, 0.02; axis 2, 0.015. See Table 1 for breed codes.
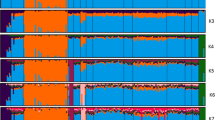
Our data set of 30 breeds was best described by K=25 genetic clusters (likelihood Ln values declined with K>25, and K=25 had a significantly higher Ln than K=24, two-tailed t-test, N1=28, N2=19, t=−2.475, P=0.017). DISTRUCT (Rosenberg, 2004) plots constructed for K=25 are shown in Figure 2a. The Mouflon, Soay, Icelandic, North Ronaldsay, Heidschnuke, Skudde, Coburg, Šumavka, Racka and Bizet remained the most genetically pure breeds (that is free from admixture) from K=23 to K=30. Clear evidence of admixture was found between two Greek breeds, with several Argos individuals having Lesvos multilocus genotypes (Figure 2a).

Individual assignment based on Bayesian cluster analysis. Plots are constructed using the program DISTRUCT (Rosenberg, 2004). The width of each segment relates to the sample size of the breed (Table 1). Individuals of different breeds are separated by black segments. Each individual is represented by a vertical line corresponding to its membership coefficient, that is a solid line corresponds to a membership coefficient of one (indicating no admixture). (a) Whole data set, K=25. Note the obvious admixture of Lesvos genotypes into Argos, the sharing of clusters between Turcana and Tsigai, Friesland and Zeeland, and Cyprus fat-tail and Chios. (b) Friesland and Zeeland investigated in more detail. The highest likelihood was found when K=3. Note the presence of two clusters in the Zeeland. (c) Cyprus fat-tail and Chios shown when K=4. Cyprus fat-tail individuals are split into several groups.
Three pairs of breeds shared clusters when K=25 (Turcana and Tsigai, Friesland and Zeeland, and Cyprus fat-tail and Chios; Figure 2a), and were, therefore, analysed independently. Turcana and Tsigai were run with K=1–6, but genetic clusters were not identified within these breeds (not shown). Friesland and Zeeland were run with K=1–5, and the highest Ln was found for K=3 (Figure 2b). Two clusters were found in the Zeeland, and all Friesland individuals except for one, which has a Zeeland genotype, were members of the same genetic cluster, whereas four Zeeland individuals have genotypes corresponding more to the Friesland (Figure 2b). Cyprus fat-tail and Chios were run with K=1–5. While these two breeds were separated into two genetic clusters when K=2, the highest likelihood was found for K=4. Under K=4, Chios remained a single cluster, whereas Cyprus fat-tail individuals were split into several groups (Figure 2c).
Discussion
Our survey of 23 microsatellite markers in European sheep breeds revealed that 87% of the variation is found within rather than among breeds. This situation mirrors other domestic species such as cattle (Wiener et al., 2004), and levels of within-breed diversity (Table 3) are also comparable to those from other domestic species, for example cattle (Katanen et al., 2000; Kim et al., 2002; Mateus et al., 2004), horses (Aberle et al., 2004) and goats (Saitbekova et al., 1999; Cañon et al., 2006). In contrast to most other domestic species however, we found extensive evidence for heterozygote deficit in European sheep breeds. Even after removing the most extreme loci in terms of null alleles, almost 30% of breed × locus comparisons still show heterozygote deficit. Other studies have also reported heterozygote deficit in domestic sheep, although generally not to the extent described here (for example, Tapio et al., 2003, 2005; Calvo et al., 2006; Peter et al., 2007; Santucci et al., 2007). For other domestic species such as cattle (refs), pigs (refs), horses (refs) and goats (refs), deviation from HW equilibrium seems to be an exception rather than a rule.
Other than the occurrence of null alleles, there are two potential explanations for this widespread heterozygote deficit in European sheep: (1) subdivision among flocks leading to a Wahlund effect and (2) nonrandom mating due to inbreeding. First, subdivision is highly likely because although flocks are not closed units, gene flow may be restricted, especially between flocks that are geographically distant. If there is subdivision among flocks, sampling several flocks will lead to positive estimates of Fis. It is possible that our sampling strategy exacerbated this effect by including a small number of individuals from more than one flock. This is particularly illustrated by the case of the wild Mouflon, which was sampled from Corsica, Sardinia and Cyprus, and has the highest Fis. Subdivision caused by geographical isolation, confounded by sampling strategy, has therefore certainly contributed to heterozygote deficit. However, breeds for which just one flock was sampled also have positive Fis values (for example, Racka, Šumavka, Aragonesa, North Ronaldsay, Soay), suggesting that subdivision does not entirely account for HW deviations.
Second, nonrandom mating due to inbreeding is also likely, since a small number of rams are typically used for breeding purposes. Interestingly, the breed with the lowest Fis (excluding those with n<15) is the Soay, the only breed that is feral and unmanaged, with the potential for random mating and only fine-scale population structuring. Low values of Fis for the Soay have been reported previously and interpreted as due to inbreeding avoidance resulting from strict female philopatry and male-biased dispersal (Coltman et al., 1999, 2003).
Despite significant heterozygote deficit, levels of heterozygosity are relatively high in commercially managed breeds compared to the primitive, feral or wild sheep such as the Californian bighorn sheep, where Ho=0.28–0.36 (Whittaker et al., 2004). A similar situation is found in domestic goats, which have substantially higher heterozygosity than their wild relatives (Saitbekova et al., 1999). This could suggest that selective breeding regimens are generally successful in maintaining diversity, but note that low diversity in wild species could be explained by successive bottleneck events, habitat fragmentation, isolation (Sage and Wolff, 1986) and strong fine-scale population structure (Coltman et al., 2003). Moreover, primitive breeds are often both genetically and geographically isolated, so genetic drift is likely to be a key factor in determining their diversity.
Although most of the genetic diversity is explained within breeds, the global estimate of Fst (13%) is quite high compared to the 7–11% reported for European cattle breeds (MacHugh et al., 1998; Katanen et al., 2000; Cañon et al., 2001; Mateus et al., 2004), or 8% for horses (Cañon et al., 2000). With the exception of only three comparisons (Turcana versus Tsigai, Thônes-Marthod versus Leccese and Thônes-Marthod versus Istrian), all breeds are significantly genetically differentiated. We identified several breeds that are very distinct and the Friesland and Soay breeds have particularly high Fst values. These breeds may harbour important disease resistance or uniquely adapted alleles and should, therefore, be given priority for conservation. Global estimates of Fis (12.3%) and Fst are similar; therefore, the variance explained by difference among breeds can be partially accounted for by correlations of allele frequencies within breeds.
Importance of geography and breed type
In the absence of recurrent introgression, we might expect most genetic diversity to be concentrated near the Near Eastern centre of domestication and then to decline with distance, since early farmers transported only a small sample of their diverse original stock (Bruford et al., 2003; Bruford, 2004). Such a pattern is suggested from analysis of sheep and cattle mitochondrial DNA sequences (Troy et al., 2001; Bruford and Townsend, 2006) and also from a recent Bayesian cluster analysis of European sheep breeds that revealed a southeast to northwest cline (Peter et al., 2007). Moreover, southern breeds have larger population sizes and have been transported more extensively than their northern counterparts (Mason, 1967). We therefore predicted higher within-breed diversity and lower genetic differentiation in southern than in northern breeds. Consistent with this prediction, the main geographical component to the partitioning of within-breed diversity is the difference between northern and southern breeds; southern breeds tend to have higher levels of heterozygosity than northern breeds (see also Peter et al., 2007), and the most distinctive breeds are all from northern Europe (Friesland, Icelandic, North Ronaldsay, Skudde and Soay). The cluster analysis also revealed more widespread evidence for admixture in southern than in northern breeds.
Domestic breeds are often categorized into ‘types’ according to morphological similarities, ecology and origins. Breeds of the same type are expected to be genetically similar, but this was generally not found to be the case. Although breed type explains the largest component of variance among groups, it only explains 2.7% of the total variation, and there is no general tendency for breeds to cluster by type in the PCA. In fact, three of the most differentiated breeds in the PCA (Soay, North Ronaldsay and Icelandic) are all of the northern short-tailed type. Similarities between breeds of the same type were, however, identified in the Bayesian cluster analysis. For example, the Dutch Friesland and Zeeland breeds, which are both of the marsh rat-tail type, always shared a cluster when K=23–30.
Application to conservation and management
According to Weitzman (1992), for a breed to become a priority for conservation, it must add new diversity elements to a pre-defined set of conservation priority breeds. Breeds that add the highest overall genetic distance to the remainder of the set should be given priority (Bruford et al., 2003). Those breeds that are clearly differentiated in our PCA (Friesland, Icelandic, North Ronaldsay, Soay and Skudde) or stand out from our cluster analysis as harbouring unique genotypes (Soay, Icelandic, North Ronaldsay, Heidschnuke, Skudde, Coburg, Šumavka, Racka and Bizet) are, therefore, obvious targets for conservation. This approach should be treated with caution though because it fails to account for the diversity and geographical structure that can be found within some breeds. It should also be noted that neutral genetic variation does not necessarily correlate with variation in phenotype or quantitative traits, therefore, planning based on microsatellite variation alone might not be sufficient. Genetic distance estimates vary according to the marker used and the recent demographic history of the breed. For instance a severely bottlenecked breed will have a large genetic distance but often little within-breed diversity relative to other breeds. The Friesland, which has been bred true to maintain its high milk productivity following bottlenecks in the 1890s and 1960s (J Lenstra, personal communication), is a good example of this, since it exhibits very low within-breed diversity but is highly differentiated from other breeds.
Our analyses illustrate the utility of Bayesian cluster analysis to identify breeds that have or have not been bred true, which is potentially useful for breed management. Clear evidence of admixture was found between two Greek breeds, which are both Zackel × fat-tail type, with introgression of Lesvos genotypes into the Argos breed. A more fine-scale analysis clearly identified admixed individuals between the Friesland and Zeeland breeds, despite the efforts of breeders to keep the Friesland completely ‘pure’.
In summary, genetic diversity in European domestic sheep breeds is characterized by extensive heterozygote deficit, most likely due to subdivision within breeds and nonrandom mating due to inbreeding. Isolation in terms of geography or breed management has been crucially important in reducing within-breed variation and augmenting among-breed differentiation. We identified breeds that possess highly distinct genotypes, indicative of long histories of isolation, a distinct origin or the effects of selection. Finally, we illustrated that Bayesian clustering methods are valuable tools for detecting unrecorded admixture among breeds. This method could be valuable when breeders wish to establish current practices ongoing in their breeds.
References
Aberle KS, Hamann H, Drogemuller C, Distl O (2004). Genetic diversity in German draught horse breeds compared with a group of primitive, riding and wild horses by means of microsatellite DNA markers. Anim Genet 35: 270–277.
Arevalo E, Holder D, Derr JN, Bhebe E, Linn RA, Taylor JF et al. (1994). Caprine microsatellite dinucleotide repeat polymorphisms at the SRCRSP1, SRCRSP2, SRCRSP3, SRCRSP4 and SRCRSP5 loci. Anim Genet 25: 202.
Arranz J, Bayon Y, Primitivo FS (1998). Genetic relationships among Spanish sheep using microsatellites. Anim Genet 29: 435–440.
Bhebhe E, Kogi J, Holder D, Arevalo E, Derr JN, Linn RA et al. (1994). Caprine microsatellite dinucleotide repeat polymorphisms at the SRCRSP6, SRCRSP7, SRCRSP8, SRCRSP9 and SRCRSP10 loci. Anim Genet 25: 203.
Bishop MD, Kappes SM, Keele JW, Stone RT, Sunden S, Hawkins G et al. (1994). A genetic linkage map for cattle. Genetics 136: 619–639.
Brezinsky L, Kemp SJ, Teale AJ (1993). Five polymorphic bovine microsatellites (ILSTS010-014). Anim Genet 24: 75–76.
Bruford MW (2004). Conservation genetics of UK livestock: from molecules to management. In: Simm G, Villanueva B, Townsend S (eds). Farm Animal Genetic Resources. Nottingham University Press: Nottingham, UK, pp 151–169.
Bruford MW, Bradley DG, Luikart G (2003). DNA markers reveal the complexity of livestock domestication. Nat Rev Genet 4: 900–910.
Bruford MW, Townsend SJ (2006). Case studies in the genetics of animal domestication: sheep. In: Zeder M, Decker-Walters D, Bradley D, Smith BD (eds). Documenting Domestication: New Genetic and Archaeological Paradigms. California University Press: Berkeley, CA, USA.
Buchanan FC, Swarbrick PA, Crawford AM (1992). Ovine dinucleotide repeat polymorphism at the MAF65 locus. Anim Genet 23: 85.
Buchanan FC, Crawford AM (1992a). Ovine dinucleotide repeat polymorphism at the Maf70 locus. Anim Genet 23: 185.
Buchanan FC, Crawford AM (1992b). Ovine dinucleotide repeat polymorphism at the Maf209 locus. Anim Genet 23: 183.
Buchanan FC, Crawford AM (1992c). Ovine dinucleotide repeat polymorphism at the Maf214 Locus. Anim Genet 23: 394.
Buchanan FC, Crawford AM (1993). Ovine microsatellites at the OarFCB11, OarFCB128, OarFCB193, OarFCB266 and OarFCB304 loci. Anim Genet 24: 145.
Buchanan FC, Galloway S, Crawford AM (1994). Ovine microsatellites at the OarFCB5, OarFCB19, OarFCB20, OarFCB48, OarFCB129, and OarFCB226 loci. Anim Genet 25: 60.
Calvo JH, Bouzada JA, Jurado JJ, Serrano M (2006). Genetic substructure of the Spanish Manchega sheep breed. Small Rumin Res 64: 116–125.
Cañon J, Alexandrino P, Bessa I, Carleos C, Carretero Y, Dunner S et al. (2001). Genetic diversity measures of local European beef cattle breeds for conservation purposes. Genet Sel Evol 33: 311–332.
Cañon J, Checa ML, Carleos C, Vega-Pla JL, Vallejo M, Dunner S (2000). The genetic structure of Spanish Celtic horse breeds inferred from microsatellite data. Anim Genet 31: 39–48.
Cañon J, Garcia D, Garcia-Atance MA, Obexer-Ruff G, Lenstra JA, Ajmone-Marsan P et al. (2006). Geographical partitioning of goat diversity in Europe and the Middle East. Anim Genet 37: 327–334.
Chakraborty R, De Andrade M, Daiger SP, Budowle B (1992). Apparent heterozygote deficiencies observed in DNA typing data and their implications in forensic applications. Ann Hum Genet 56: 45–57.
Chapuis M-P, Estoup A (2007). Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24: 621–631.
Coltman DW, Pilkington JG, Pemberton JM (2003). Fine-scale genetic structure in a free-living ungulate population. Mol Ecol 12: 733–742.
Coltman DW, Pilkington JG, Smith JA, Pemberton JM (1999). Parasite-mediated selection against inbred Soay sheep in a free-living, island population. Evolution 53: 1259–1267.
Crawford A, Dodds K, Ede A, Pierson CA, Montgomery GW, Garmonsway HG et al. (1995). An autosomal genetic linkage map of the sheep genome. Genetics 140: 703–724.
Dakin EE, Avise JC (2004). Microsatellite null alleles in parentage analysis. Heredity 93: 504–509.
Dempster AP, Laird NM, Rubin DB (1977). Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc B 39: 1–38.
Diez-Tascon C, Littlejohn RP, Almeida PAR, Crawford AM (2000). Genetic variation within the Merino sheep breed: analysis of closely related populations using microsatellites. Anim Genet 31: 243–251.
El Mousadik A, Petit RJ (1996). High level of genetic differentiation for allelic richness among populations of the argan tree Argania spinosa (L.) skeels endemic to Morocco. Theor Appl Genet 92: 832–839.
Excoffier L, Smouse PE, Quattro JM (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
FAO (2000). Domestic Animal Diversity Information System (DAD-IS 2.0):http://www.fao.org/dad-is/.
Goudet J (2000). FSTAT, a programme to estimate and test gene diversities and fixation indices (version 2.9.1). Available from http://www.unil.ch/izea/softwares/fstat.html (updated from Goudet 1995).
Guo J, Du LX, Ma YH, Guan WJ, Li HB, Zhao QJ et al. (2005). A novel maternal lineage revealed in sheep (Ovis aries). Anim Genet 36: 331–336.
Guo S, Thompson EA (1992). Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometrics 48: 361–372.
Hiendleder S, Kaupe B, Wassmuth R, Janke A (2002). Molecular analysis of wild and domestic sheep questions current nomenclature and provides evidence for domestication from two different subspecies. Proc R Soc London B 269: 893–904.
Hiendleder S, Mainz K, Plante Y, Lewalski H (1998). Analysis of mitochondrial DNA indicates that domestic sheep are derived from two different ancestral maternal sources: no evidence for contributions from Urial and Argali sheep. J Hered 89: 113–120.
Hoelzel AR, Dover G (1991). Molecular Genetic Ecology. Information Press: Oxford.
Katanen J, Olsaker I, Holm LE, Lien S, Vilkki J, Brusgaard K et al. (2000). Genetic diversity and population structure of 20 north European cattle breeds. J Hered 91: 446–457.
Kemp S, Hishida O, Wambugu J, Rink A, Longeri ML, Ma RZ et al. (1995). A panel of polymorphic bovine, ovine and caprine microsatellite markers. Anim Genet 26: 299–306.
Kim KS, Yeo JS, Choi CB (2002). Genetic diversity of north-east Asian cattle based on microsatellite data. Anim Genet 33: 201–204.
MacHugh D, Loftus RT, Cunningham P, Bradley DG (1998). Genetic structure of seven European cattle breeds assessed using 20 microsatellite markers. Anim Genet 29: 333–340.
Martín-Burriel I, Garcia-Muro E, Zargoza P (1999). Genetic diversity analysis of six Spanish native cattle breeds using microsatellites. Anim Genet 30: 177–182.
Martinez AM, Delgado JV, Rodero A, Vega-Pla JL (2000). Genetic structure of the Iberian pig breed using microsatellites. Anim Genet 31: 295–301.
Mason I (1967). Sheep Breeds of the Mediterranean. CAB: Slough, UK.
Mateus JC, Penedo MCT, Alves VC, Ramos M, Rangel-Figueiredo T (2004). Genetic diversity and differentiation in Portuguese cattle breeds using microsatellites. Anim Genet 35: 106–113.
Mendelsohn R (2003). The challenge of conserving indigenous domesticated animals. Ecol Econ 45: 501–510.
Paetkau D, Strobeck C (1995). The molecular basis and evolutionary history of a microsatellite null allele in bears. Mol Ecol 4: 519–520.
Pariset L, Savarese MC, Cappuccio I, Valentini A (2003). Use of microsatellites for genetic variation and inbreeding analysis in Sarda sheep flocks of central Italy. J Anim Breed Genet 120: 425–432.
Pedrosa S, Uzun M, Arranz JJ, Gutierrez-Gill B, Primitivo FS, Bayon Y (2005). Evidence of three maternal lineages in near eastern sheep supporting multiple domestication events. Proc R Soc London B 272: 2211–2217.
Peter C, Bruford MW, Perez T, Dalamitra S, Hewitt GM, Erhardt G, The Econogene Consortium (2007). Genetic diversity and subdivision of 57 European and Middle-Eastern sheep breeds. Anim Genet 38: 37–44.
Peter C, Prinzenberg EM, Erhardt G, The Econogene Consortium (2005). Null allele at the OarAE129 locus and corresponding allele frequencies in German sheep breeds. Anim Genet 36: 92.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Raymond M, Rousset F (1995). GENEPOP (version 1.2) population genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Rege JEO, Gibson JP (2003). Animal genetic resources and economic development: issues in relation to economic valuation. Ecol Econ 45: 319–330.
Rosenberg NA (2004). DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4: 137–138.
Rousset F, Raymond M (1995). Testing heterozygote excess and deficiency. Genetics 140: 1413–1419.
Ryder M (1981). Sheep. In: Mason I (ed). Evolution of Domesticated Animals. Longman: London and New York, pp 63–85.
Sage RD, Wolff JO (1986). Pleistocene glaciations, fluctuating ranges, and low genetic-variability in a large mammal (Ovis dalli). Evolution 40: 1092–1095.
Saitbekova N, Gaillard C, Obexer-Ruff G, Dolf G (1999). Genetic diversity in Swiss goat breeds based on microsatellite analysis. Anim Genet 30: 36–41.
Sambrook J, Fritsch EF, Maniatis T (1989). Molecular Cloning: a Laboratory Manual. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY.
Santucci F, Ibrahim KM, Bruzzone A, Hewitt GM (2007). Selection on MHC-linked microsatellite loci in sheep populations. Heredity [E-pub ahead of print, 23 July 2007; http://dx.doi.org/10.1038/sj.hdy.6801006].
Schneider S, Roessli D, Excoffier L (2000). ARLEQUIN ver. 2.000: a Software for Population Genetic Data Analysis. Genetics and Biometry Laboratory, University of Geneva: Switzerland.
Scott P, Densmore LL (1991). Mitochondrial DNA isolation. In: Hoelzel AR (ed). Molecular Genetic Analysis of Populations: a Practical Approach. IRL Press: Oxford, UK, pp 29–58.
Signorello G, Pappalardo G (2003). Domestic animal biodiversity conservation: a case study of rural development plans in the European Union. Ecol Econ 45: 487–499.
Slatkin M (1995). A measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457–462.
Steffen P, Eggen A, Dietz A, Womack JE, Stranzinger G, Fries R (1993). Isolation and mapping of polymorphic microsatellites in cattle. Anim Genet 24: 121–124.
Tapio M, Miceikiene I, Vilkki J, Kantanen J (2003). Comparison of microsatellite and blood protein diversity in sheep: inconsistencies in fragmented breeds. Mol Ecol 12: 2045–2056.
Tapio M, Tapio I, Grislis Z, Holm LE, Jeppsson S, Kantanen J et al. (2005). Native breeds demonstrate high contributions to the molecular variation in northern European sheep. Mol Ecol 14: 3951–3963.
Troy CS, MacHugh DE, Bailey JF, Magee DA, Loftus RT, Cunningham P et al. (2001). Genetic evidence for near-eastern origins of European cattle. Nature 410: 1088–1091.
Vaiman D, Mercier D, Moazami-Goudarzi K, Eggen A, Ciampolini R, Lepingle A et al. (1994). A set of 99 cattle microsatellites: characterisation, synteny mapping, and polymorphism. Mamm Genom 5: 288–297.
Walsh P, Metzger DA, Higuchi R (1991). Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10: 506–513.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Weitzman ML (1992). On diversity. Quart J Econ CVII: 363–405.
Whittaker DG, Osterman SD, Boyce WM (2004). Genetic variability of reintroduced California bighorn sheep in Oregon. J Wildlife Manage 68: 850–859.
Wiener P, Burton D, Williams JL (2004). Breed relationships and definition in British cattle: a genetic analysis. Heredity 93: 597–602.
Wright S (1951). The genetic structure of populations. Ann Eugen 15: 323–354.
Wright S (1977). Evolution and the Genetics of Populations. Vol. 3: Experimental Results and Evolutionary Deductions. University of Chicago Press: Chicago.
Acknowledgements
This work was funded by the European Commission under contract BIO4961189 in the Biotech 2 programme of the Fourth Framework programme. The views expressed in this article do not represent those of the European Commission. We thank all farmers and collaborators who provided the many samples described in this study, including J Lenstra, LH Alderson, A Valentini, J Pemberton, A Knowles, A Reeb, Athalassa ARI Farm, M Ferrano, F Rosatini, M Albrecht, V Tauber, R Falge, M Reichel, L Head, D Ridd, G Leeves, O Dyrmundsson, M Hughes, I Bodo, C Ehling, F Pilla, A Alberti, GM Levrino, C Papachristoforou, E Rogdakis, P Vellema and J Futerova. LLH also thanks François Balloux and Jerome Goudet for helpful discussion.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary information
Rights and permissions
About this article
Cite this article
Lawson Handley, LJ., Byrne, K., Santucci, F. et al. Genetic structure of European sheep breeds. Heredity 99, 620–631 (2007). https://doi.org/10.1038/sj.hdy.6801039
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6801039
Keywords
This article is cited by
-
Runs of homozygosity and cross-generational inbreeding of Iranian fat-tailed sheep
Heredity (2023)
-
Origin, demographics, inbreeding, phylogenetics, and phenogenetics of Karamaniko breed, a major common ancestor of the autochthonous Greek sheep
Tropical Animal Health and Production (2022)
-
RNA-Seq based genetic variant discovery provides new insights into controlling fat deposition in the tail of sheep
Scientific Reports (2020)
-
Retroviral analysis reveals the ancient origin of Kihnu native sheep in Estonia: implications for breed conservation
Scientific Reports (2020)
-
Approaching sheep herds origins and the emergence of the wool economy in continental Europe during the Bronze Age
Archaeological and Anthropological Sciences (2019)
