
Abstract
Smith–Magenis syndrome (SMS) is a complex neurobehavioral disorder caused by haploinsufficiency of the retinoic acid-induced 1 (RAI1) gene on chromosome 17p11.2. Diagnostic strategies include molecular identification of a 17p11.2 microdeletion encompassing RAI1 or a mutation in RAI1. G-banding and fluorescent in situ hybridization (FISH) are the classical methods used to detect the SMS deletions, while multiplex ligation-dependent probe amplification (MLPA) and real-time quantitative PCR are the newer, cost-effective, and high-throughput technologies. Most SMS features are due to RAI1 haploinsufficiency, while the variability and severity of the disorder are modified by other genes in the 17p11.2 region. The functional role for RAI1 is not completely understood, but it is likely involved in transcription, based on homology and preliminary studies. Management of SMS is primarily a multidisciplinary approach and involves treatment for sleep disturbance, speech and occupational therapies, minor medical interventions, and management of behaviors.
Similar content being viewed by others


In brief
-
Smith–Magenis syndrome is a multiple congenital anomalies/mental retardation disorder caused by an interstitial deletion involving chromosome 17p11.2 containing the retinoic acid-induced 1 (RAI1) gene or due to mutation of RAI1.
-
Typically a sporadic disorder with an estimated prevalence of 1:15 000–25 000.
-
SMS patients have mental retardation, distinctive behavioral features, craniofacial and skeletal anomalies, speech and developmental delay, and sleep disturbance.
-
Hypotonia, hearing loss and chronic ear infections, eye abnormalities, systemic features such as cardiac and renal defects, and, occasionally, cleft lip and/or palate is also observed.
-
Approximately 90% of SMS cases have a fluorescent in situ hybridization (FISH) detectable 17p11.2 microdeletion (ranging from 1.5 to 9 Mb), while the remaining 10% have a mutation in RAI1.
-
Approximately 70% of patients with 17p11.2 deletions have a ‘common’ ∼3.7-Mb deletion, whereas the remaining 30% have larger or smaller deletions.
-
Management includes therapy for sleep disturbance (acebutolol and melatonin), early childhood intervention programs, special education and vocational training, and multidisciplinary evaluation for behavioral and systemic manifestations.
-
Recurrence risk for sibs of the proband, if the parental chromosome/gene analyses are normal, is less than 1%. Risk increases if a parent of the proband carries a balanced chromosomal rearrangement or if mosaicism for either a deletion or RAI1 mutation is present in either parent. Mosaicism in a parent of an affected child is estimated at 3–5%.
Introduction
Smith–Magenis syndrome (SMS; OMIM #182290, *607642) is a complex disorder characterized by variable mental retardation, sleep disturbance, craniofacial and skeletal anomalies, self-injurious and attention-seeking behaviors, and speech and motor delay.1 SMS is generally a sporadic disorder caused by either a 17p11.2 deletion encompassing the retinoic acid-induced 1 (RAI1) gene or a mutation of RAI1.1, 2, 3 Although the incidence of SMS is estimated to be ∼1:15 000–25 000, it is often underdiagnosed.4
Clinical overview
Smith–Magenis syndrome is characterized by a clinically recognizable phenotype that includes physical, developmental, neurological, and behavioral features. Physical features consist of distinctive craniofacial anomalies including brachycephaly, frontal bossing, hypertelorism, synophrys, upslanting palpebral fissures, midface hypoplasia, a broad square-shaped face with depressed nasal bridge, an everted upper lip with a ‘tented’ appearance (cupid's bow upper lip), and micrognathia in infancy progressing toward relative prognathism with age (Figures 1 and 2; Supplementary Figure S1; Table 1).14 Additional SMS case photos ranging in age from infant to adult are provided in Supplementary Figure S1. Dental anomalies include tooth agenesis, especially of premolars, and taurodontism.5 Less frequently, cleft lip and/or palate is also reported.1 Short stature (<5th percentile) was observed in ∼67% of young patients, while 33% were overweight in an evaluation of 105 SMS cases at various ages (Table 1).6 Short stature resolves over time with most individuals reaching the 10–25th percentile by adulthood.15 Obesity in teens and adults is common, typically with broad chests and truncal obesity. Other skeletal anomalies include brachydactyly, scoliosis, fifth-finger clinodactyly, 2/3 toe syndactyly, forearm and elbow limitations, vertebral anomalies, persistent fetal finger pads, and polydactyly (Figure 2; Table 1).1, 7, 16, 17, 18 Otolaryngological problems such as hearing loss, velopharyngeal insufficiency, a hoarse deep voice, and vocal cord nodules and polyps are also common (Table 1).7, 19, 20 Hearing loss, present in 60% of SMS patients, is variable and may be mild to moderate (conductive) or severe (sensorineural) and is often associated with chronic ear infections (Table 1).4, 7, 19 Ophthalmologic features are present in >60% of SMS patients and include myopia, iris anomalies such as heterochromic irides or Wolfflin–Kruckmann spots (iris hamartomas), strabismus, microcornea, and, rarely, retinal detachment (often resulting from violent behaviors).8, 21

An algorithm for the diagnosis of SMS is shown. *Possible other diagnoses include 9q34 deletion syndrome, Prader–Willi syndrome, 22q11.2 deletion syndrome, Williams syndrome, and/or Sotos syndrome. Please refer to Table 1 also.

Age progression of female subjects with SMS. Typical SMS infant phenotype with ‘tented’ upper lip and depressed nasal bridge at birth (a), at age 4 months (b), and with hand-clasping behavior at age 1 year (c) is shown. A white patch on the dorsum of left forearm due to skin picking is illustrated in a toddler (also hand clasping) at age 2 years (d). The same individual at ages 13 years (e) and 20 years (f), and photos at 21 years of age illustrating open wounds and scarring on forearms from skin picking (g), brachydactyly and nail-yanking lesions on hands (h), and feet with brachydactyly and nails damaged from nail yanking (i).
Prenatal history of SMS is notable for significantly decreased fetal movement in 50% of pregnancies.22 The infant with SMS is usually born at term, initially with normal anthropometry, later progressing toward greater craniofacial width than depth or height. Early infancy is complicated by feeding difficulties leading to failure to thrive, marked oral sensory motor dysfunction with poor suckling reflex, gastroesophageal reflux, and hypotonia.22 Infantile hypotonia accompanied by generalized lethargy mimics Down syndrome and Prader–Willi syndrome phenotypes (Table 2).23
Most SMS individuals have mild-to-moderate mental retardation with IQ ranging between 20–78.7 School-age children with IQs in the low normal range have been identified; however, IQ decreases as the child ages, ultimately placing the individual in the mild mental retardation range by adulthood (SH Elsea, unpublished data). Individuals with SMS also have decreased ability in sequential processing and short-term memory, with stronger long-term memory and perceptual closure.7 Delayed speech with or without hearing loss occurs in ∼96% of SMS patients. Individuals with SMS have better receptive language skills than expressive language, while their social/emotional functions remain within the normal range.22, 23 Many of the children will have significant speech delay up to 7 years of age, and thus are often diagnosed as autistic but once speaking, are very interactive and communicative. In addition, delayed fine/gross motor skills, problems with sensory integration, and poor adaptive function are seen. Other neurological features include pes cavus or pes planus, an abnormal ‘festinating’ gait, balance problems, and a decreased sensitivity to pain, which is often observed in association with self-injury in this disorder.7 Peripheral neuropathy is a typical finding in SMS but is not associated with abnormal nerve conduction velocities; however, patients with large deletions encompassing PMP22 gene have focal pressure neuropathies including carpal tunnel syndrome and peroneal palsy, polyneuropathy, decreased reflexes, and reduced nerve conduction velocity, consistent with HNPP (Table 3; Figure 3).4, 24

Map of the SMS region. A schematic showing the SMS region with representative genes and proximal, middle, and distal repeats. The different-sized 17p11.2 deletions identified in SMS cases including common, large, small, and atypical deletions are shown. Commercially available FISH probes useful for SMS diagnosis are also represented. Map not drawn to scale.
Sleep disturbance has been reported in 75–100% of SMS cases and is one of the earliest diagnostic indicators of SMS (Table 1).6, 7, 9, 25 Infants typically experienced hypersomnolence during the first year of life.22 Sleep disturbances in older children include difficulties falling asleep, diminished REM sleep, reduced 24-h and night sleep, fragmented and shortened sleep cycles with frequent nocturnal and early-morning awakenings, and excessive daytime sleepiness. These abnormal sleep patterns are due to an inverted circadian rhythm of melatonin.26, 27 Aberrant melatonin synthesis/degradation pathway has been proposed to be the underlying cause for the inverted circadian rhythm.27
Behavioral issues are one of the unique characteristic features of SMS (Table 1; Supplementary Figure S2). Maladaptive behaviors are a cause of major concern and include frequent outbursts/temper tantrums, attention seeking, aggression, disobedience, distraction, and self-injurious behaviors.10 Self-injurious behaviors, including head banging, skin picking, and wrist biting often begin at 15–18 months of age, while two features unique to SMS, onychotillomania (pulling out of fingernails and toenails) and polyembolokoilamania (insertion of objects into bodily orifices), are more often seen in older children (Figure 2).4, 28 Stereotypical behaviors unique to SMS include the spasmodic upper body squeeze or ‘self-hugging,’ and page-flipping or ‘lick and flip’ behavior often seen in association with excitement (Supplementary Figure S2).29 Additionally, insertion of a hand in the mouth, teeth grinding, body rocking, and spinning or twirling objects are documented.10, 28 The behavioral phenotype of SMS escalates with age, typically with the onset of puberty. Age, degree of developmental delay, severity of any associated systemic disorder, and degree of sleep disturbance have a strong influence on maladaptive behaviors.10 Individuals also have a lack of respect for personal space during a conversation and are emotionally volatile. These behaviors are present in all persons with SMS, but specific behaviors may change over time, with higher functioning individuals having a broader repertoire of behaviors, particularly evident in those persons with RAI1 mutations.
Organ malformations, present in 30–40% of SMS patients, include cardiac, renal, and CNS abnormalities (Table 1). Cardiac findings such as ventricular septal defect, atrial septal defect, tricuspid stenosis, mitral stenosis, tricuspid and mitral regurgitation, aortic stenosis, pulmonary stenosis, mitral valve prolapse, tetralogy of Fallot, and total anomalous pulmonary venous return are seen in 30% of SMS individuals.6 Enlarged kidneys, duplication of the collecting duct, ectopic kidneys, and unilateral kidney agenesis have also been documented.6, 7 Malformations observed by brain imaging include ventriculomegaly, enlarged cisterna magna, partial agenesis of cerebellar vermis, frontal lobe calcification, and Joubert syndrome.1, 7, 30 Similarly, MRI and PET studies on SMS patients have shown decreased gray matter in the insula and lenticular nucleus.31 Recently, Girirajan et al24 reported an SMS case with moyamoya disease (occlusion of the circle of Willis). Seizures, diagnosed clinically as well as by electroencephalogram evaluations, are present in 10–30% of patients and may be variable in age of onset, type, and severity; frequent occurrence of catamenial seizures in female patients is also worth noting.6, 7 Decreased levels of immunoglobulins (IgA, IgE, and IgG) and thyroxine deficiency have also been reported, and levels should be evaluated in all cases.7 Smith et al32 reported hypercholesterolemia in 57% of an SMS cohort.
Diagnostic approaches
The diagnosis of SMS is based upon initial clinical suspicion of the disorder followed by a molecular confirmation of the chromosomal/gene defect. A list of differential diagnoses for SMS is given in Table 2. Clinical acumen to identify SMS ‘facial gestalt,’ careful history-taking for birth defects, sleep disturbance, delayed milestones, chronic ear infections, self-injurious behaviors (Supplementary Figure S2), and family history are important to recognize the characteristic features. In addition to behavioral features unique to SMS, unusual findings typically observed that may be clues to the diagnosis include a hoarse, harsh, guttural voice, characteristic feet and hands (Figure 2), truncal obesity with onset in teenage or early adult years often with thin legs, excellent long-term memory, good ability and focus with computers, and scarring and/or infection from repeated skin picking (Figure 2).
Molecular confirmation of SMS (see Figures 1 and 3) is done by detection of a 17p11.2 microdeletion using any of a plethora of cytogenetic and molecular techniques that differ in throughput, specificity, and cost-efficiencies, and include G-banded cytogenetic analysis (550 band or higher), fluorescent in situ hybridization (FISH) using an RAI1-specific probe, multiplex ligation-dependent probe amplification (MLPA), real-time quantitative PCR (qPCR), and targeted chromosome microarrays for comparative genomic hybridization (CGH).24, 33 While FISH and G-banding are classically used for SMS diagnosis in a clinical cytogenetic laboratory, MLPA and qPCR are newer, cost-efficient methodologies for rapid, high-throughput diagnosis that require only DNA for analysis.34 Further, MLPA and real-time qPCR can identify smaller deletions at a higher resolution, usually missed by FISH or G-banding, such as exonic deletions involving RAI1 (Figure 3).24 Whole genome chromosome microarray studies (CGH) will also identify 17p11.2 deletions. Individuals in whom no 17p11.2 deletion can be found should have the RAI1 gene sequenced to detect heterozygous nucleotide variations (Figure 1; Table 4). Upon detection of a nucleotide change, parental samples are evaluated to confirm the mutation is de novo (Figure 1). So far, only one case of SMS with an inherited RAI1 nucleotide change has been reported; however, parental mosaicism for an RAI1 mutation has been documented (SH Elsea, unpublished results).2, 37 With the benefit of retrospection, it is prudent to consider evaluation of all parents of SMS cases with RAI1 mutations for familial mutations or mosaicism. The presence of an inherited mutation or mosaicism in the parent of a child with SMS would greatly alter the recurrence risks. Further, the identification of familial, population-specific, ‘novel’ single-nucleotide polymorphisms complicates diagnosis. The diagnostic strategy for SMS is illustrated in Figure 1.
Molecular and genetic basis of disease
Mechanism of SMS deletions
Approximately 90% of all reported cases with SMS have a 17p11.2 deletion, while the remaining 10% have a mutation in the RAI1 gene. Microdeletions, including 17p11.2 SMS deletions, are caused by irregularities in the chromosomal recombination mechanism sponsored by repeat elements in the susceptible region of the genome.38 Chromosome 17p11.2p12 is one of the most recombination-prone regions of the genome and is also associated with, in addition to SMS, hereditary neuropathy with liability to pressure palsies (HNPP) and Charcot–Marie–Tooth disease type 1A (Figure 3; Table 3). Chen et al11 identified three copies of a low-copy number repeats (LCRs) flanking the SMS common deletion region. These repeats (proximal, middle, and distal SMS REPs) form substrates for inter- and intrachromosomal recombination (Figure 3). Unequal meiotic crossovers mediated through nonallelic homologous recombination occur between the proximal and distal SMS REPs in ∼70% of SMS deletion cases resulting in a common deletion (Figure 3).39 Similarly, uncommon deletions (seen in ∼30% of deletion cases) are either due to alternate LCRs, such as AT-rich repeats and Alu elements acting as homologous recombination substrates, or due to nonhomologous mechanisms.3, 40, 41 Also, no imprinting or parent-of-origin bias for the SMS deletion has been identified.42
Identification of retinoic acid-induced 1 (RAI1): the primary gene for SMS
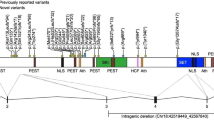
Analysis of different-sized SMS deletions revealed a common region of overlap or a ‘critical interval’ of ∼1.5 Mb within 17p11.2 (Figure 3). A contiguous physical and transcription map of the ‘critical interval’ identified 13 known genes, 14 ESTs, and 6 genomic markers.43 Sequencing of candidate genes within the ‘critical interval’ in three patients with SMS clinical features but without a 17p11.2 deletion, identified frame-shift mutations in the retinoic acid-induced 1 (RAI1) gene.2 Since then, >12 mutations including nonsense mutations, single-to-multiple nucleotide deletions, and missense mutations have been identified in RAI1 (Table 4).9, 35, 36 All mutations so far identified are located within the large exon 3 and affect all known transcripts (Figure 4). In addition, numerous single-nucleotide polymorphisms including variable polyglutamine repeats have been identified.9, 36 All reported mutations and polymorphisms are shown in Table 4.

Diagram of RAI1. The top panel shows genomic structure of RAI1 (isoform A) with six exons. All RAI1 mutations reported so far localize to exon 3, which comprises >95% of the coding sequence. Two other RAI1 isoforms (B and C) are known but not well characterized. The bottom panel illustrates the protein structure of RAI1, including the polyglutamine (Poly-Q) and polyserine (Poly-S) tracts, the bipartite nuclear localization signals (NLS), and the C-terminal PHD (plant homeodomain).
The primary transcript for RAI1 (GenBank AY172136, AJ271790; NM_030665.3; NP_109590.3; OMIM*607642) is formed by six exons generating an ∼8.5-kb mRNA and a 1906-amino-acid protein (Figure 4).44 The RAI1 protein contains a bipartite nuclear localization signal, polyglutamine and polyserine tracts, and a C-terminal plant homeodomain (PHD)/zinc-finger domain.44 The polyserine signal is similar to that found in the DRPLA and the Drosophila hairless genes, both of which are involved in neuronal development.45, 46 The polyglutamine and polyserine stretches have also been previously shown to be involved in transcription in other genes.47 The PHD domain in RAI1 is similar to that in the trithorax family of nuclear proteins, which is involved in chromatin remodeling and transcriptional regulation.48 Further, amino-acid sequence motifs representing these four domains of the RAI1 protein are similar to the transcription factor stromelysin-1 platelet-derived growth factor-responsive element-binding protein, TCF20.49, 50 Thus, it is likely that RAI1 functions in transcriptional machinery involved in growth and neurobehavioral regulation.
Genotype–phenotype correlation
All SMS patients with a 17p11.2 deletion are deleted for RAI1, and mutations in RAI1 likely result in a truncated and/or nonfunctional protein, thus resulting in haploinsufficiency.2, 35
Recently, the phenotypic role of RAI1 and the contributions of other genes in the 17p11.2 region toward SMS phenotype were evaluated by a genotype–phenotype correlation.6, 9 While RAI1 was shown to be responsible for most SMS features, other genes in the 17p11.2 region contribute to the variability and severity of the phenotype in 17p11.2 deletion cases (Table 1).9 A list of important genes mapping to 17p11.2 and their associated disorders are given in Table 3. While genes for autosomal dominant disorders are localized within this region, a 17p11.2 deletion may also unmask recessive alleles within the region, including MYO15A and ATPAF2 (Table 3), contributing to the variability of the phenotype. Short stature, hypotonia, speech and motor delay, hearing loss, frequent ear infections, and cardiac and renal defects are associated with patients with deletions, suggesting a minor role for RAI1 in these clinical features (Table 1). Thus, other genes in 17p11.2 likely contribute to these findings in 17p11.2 deletion cases. In addition, patients with RAI1 mutations may have less severe motor delay and higher functioning. They are also more likely to exhibit overeating/obesity and overgrowth phenotypes (>90th percentile for weight and height), polyembolokoilamania, self hugging, muscle cramping, and dry skin compared with patients with deletions. However, all RAI1 mutation cases so far described are phenotypically quite similar, and a bias in ascertainment must be considered; thus, the full spectrum of phenotypic effects of RAI1 mutation are not yet known.
Phenotypes in cases with small deletions are similar to those with RAI1 mutations. Edelman et al6 also reported that individuals with small deletions are less likely to show brachycephaly, dental anomalies, iris abnormalities, head banging, and hyperactivity. Incidence of behavioral features are considerably lower in patients with large and atypical deletions, most likely due to the severe, movement-limiting phenotypes including severe mental retardation and significant motor delays.9 Potential gender differences are also seen in SMS, with female patients more likely to report myopia, eating/appetite disorders, cold extremities, and problems with communication and language compared with male patients.6 The frequency of SMS features in patients with mutations compared with deletions is shown in Table 1.
Management
Appropriate assessment of the degree of cognitive, developmental, and behavioral deficits and severity of systemic/organ abnormality is essential for appropriate and specific management of SMS. During early childhood, focus should be on speech and language pathologies as well as on identifying and treating feeding problems. Speech and language evaluation is necessary for optimizing functional communication and development of intervention strategies. Early use of sign language as an adjunct to speech therapy is effective in overall speech development and also helps to decrease the child's frustrations associated with problems in expressive language, and thus, has a positive implication on behavior.7 Behavioral therapies include special education techniques; for example, children with SMS tend to do better in calmer, smaller, and more focused classroom settings. Also, emphasis on individualized instruction, structure, and routine helps minimize behavioral outbursts in the school setting. In addition, an organic cause for behaviors such as acute otitis media, gastroesophageal reflux, abdominal or joint pains, subclinical seizures, and upper respiratory tract infections should be explored and treated. Empirical evidence exists for the use of psychotropic medications (eg, thioridazine, carbamazepine, and serotonin-reuptake inhibitors) to increase attention, decrease hyperactivity, and stabilize behavior, although no single regimen has shown consistent efficacy.7 β-Blockers and mood stabilizers, such as lithium and fluoxetine, have been tried for self-injurious and aggressive behaviors with varying results.22 β-Blockers have given some positive results due to its inhibitory effects on melatonin pathway.51 Polypharmacy is typical in children with SMS as one single drug is not effective to control all the symptoms. It is, therefore, important to monitor the potential cumulative side effects of these medications and the efficacy for the individual.
A careful neurological evaluation including electroencephalography should be performed in all individuals to assess for subclinical seizures. This can be accompanied by neuroimaging to identify any anatomical brain defects. Medications should be carefully prescribed anticipating potential interactions with other behavior-modifying drugs.
Management of sleep disturbances has been one of the challenging tasks. No well-controlled treatment plan has been reported. A single uncontrolled study of SMS cases treated with oral β1-antagonist (acebutolol 10 mg/kg/day) showed suppression of daytime melatonin peaks and subjectively improved behavior. This initial study did not restore nocturnal plasma concentration of melatonin.51 Further studies showed that administration of acebutolol (10 mg/kg administered at 0800 hours) to reduce daytime melatonin secretion in combination with an evening oral dose of control-release melatonin (6 mg at 2000 hours) to restore nocturnal plasma melatonin levels caused considerable, subjectively improved behavior.52
Owing to multisystem involvement and variable presentation of SMS, a thorough clinical and diagnostic evaluation should be carried out for effective management. Annual ophthalmologic testing with corrective lenses and otolaryngological evaluations followed by pressure-equalizing tubes or hearing aids are indicated, as needed. Initial cardiac and renal work-ups consisting of echocardiogram and renal ultrasound, and periodic testing for hypercholesterolemia, immunological problems, and hormonal levels are also recommended. Family psychosocial support and counseling, including respite services, are also indicated for a better quality of life.
Conclusions
The key to a proper diagnosis and management of SMS involves correct clinical diagnosis, which may be difficult at times due to the variable presentation of the disorder, the use of rapid, high-throughput, and cost-effective molecular diagnostic tools, and timely management of clinical features and complications of the disorder. A multidisciplinary approach consisting of clinical geneticists, pediatricians, genetic counselors, molecular biologists, psychologists, dentists, speech therapists, physical therapists, and other allied health-care providers is required for a complete evaluation, understanding, and management of the disorder. While the use of MLPA and real-time qPCR for a rapid, cost-effective diagnosis is already underway in many research laboratories and in Europe, most clinical and diagnostic labs in the United States are yet to adopt these technologies. Further, use of customized array CGH is promising.
The functional role of RAI1 is not completely understood, although it is likely involved in transcription. Heterozygous mice deficient for Rai1 have obesity, craniofacial anomalies, and behavioral and neurological deficits.53 While functional abrogation of RAI1 due to a deletion or nonsense/frame-shift mutation is a likely mechanism for RAI1 haploinsufficiency, the causative role of missense mutations and the effect(s) of polymorphisms are not completely understood. The few reported cases with different types of RAI1 mutations have precluded genotype–phenotype correlations within this cohort.
While the number of SMS clinical cases diagnosed by molecular techniques is increasing, a cohort of individuals with SMS features but without a 17p11.2 deletion or a mutation in RAI1 is also emerging. Identification of RAI1-interacting genes and/or genes elsewhere in the genome that might contribute to an ‘SMS-like’ phenotype is important for a complete understanding of the pathway involved in craniofacial development, sleep, and behavior.
Accession codes
References
Smith AC, McGavran L, Robinson J et al: Interstitial deletion of (17)(p11.2p11.2) in nine patients. Am J Med Genet 1986; 24: 393–414.
Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH : Mutations in RAI1 associated with Smith–Magenis syndrome. Nat Genet 2003; 33: 466–468.
Vlangos CN, Yim DK, Elsea SH : Refinement of the Smith–Magenis syndrome critical region to approximately 950kb and assessment of 17p11.2 deletions. Are all deletions created equally? Mol Genet Metab 2003; 79: 134–141.
Greenberg F, Guzzetta V, Montes de Oca-Luna R et al: Molecular analysis of the Smith–Magenis syndrome: a possible contiguous-gene syndrome associated with del(17)(p11.2). Am J Hum Genet 1991; 49: 1207–1218.
Tomona N, Smith AC, Guadagnini JP, Hart TC : Craniofacial and dental phenotype of Smith–Magenis syndrome. Am J Med Genet A 2006; 140: 2556–2561.
Edelman E, Girirajan S, Finucane B et al: Gender, genotype, and phenotype differences in Smith–Magenis syndrome: a meta-analysis of 105 cases. Clin Genet 2007; 71: 540–550.
Greenberg F, Lewis RA, Potocki L et al: Multi-disciplinary clinical study of Smith–Magenis syndrome (deletion 17p11.2). Am J Med Genet 1996; 62: 247–254.
Chen RM, Lupski JR, Greenberg F, Lewis RA : Ophthalmic manifestations of Smith–Magenis syndrome. Ophthalmology 1996; 103: 1084–1091.
Girirajan S, Vlangos CN, Szomju BB et al: Genotype–phenotype correlation in Smith–Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum. Genet Med 2006; 8: 417–427.
Dykens EM, Smith AC : Distinctiveness and correlates of maladaptive behaviour in children and adolescents with Smith–Magenis syndrome. J Intellect Disabil Res 1998; 42 (Part 6): 481–489.
Chen KS, Manian P, Koeuth T et al: Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet 1997; 17: 154–163.
Finucane B, Dirrigl KH, Simon EW : Characterization of self-injurious behaviors in children and adults with Smith–Magenis syndrome. Am J Ment Retard 2001; 106: 52–58.
Juyal RC, Greenberg F, Mengden GA et al: Smith–Magenis syndrome deletion: a case with equivocal cytogenetic findings resolved by fluorescence in situ hybridization. Am J Med Genet 1995; 58: 286–291.
Allanson JE, Greenberg F, Smith AC : The face of Smith–Magenis syndrome: a subjective and objective study. J Med Genet 1999; 36: 394–397.
Smith ACM, Leonard AK, Gropman A, Krasnewich D : Growth assessment of Smith–Magenis syndrome, 54th Annual meeting of the American Society of Human Genetics, p.145. Toronto 2004.
Kondo I, Matsuura S, Kuwajima K et al: Diagnostic hand anomalies in Smith–Magenis syndrome: four new patients with del (17)(p11.2p11.2). Am J Med Genet 1991; 41: 225–229.
Mariannejensen L, Kirchhoff M : Polydactyly in a boy with Smith–Magenis syndrome. Clin Dysmorphol 2005; 14: 189–190.
Schlesinger AE, Potocki L, Poznanski AK, Lupski JR : The hand in Smith–Magenis syndrome (deletion 17p11.2): evaluation by metacarpophalangeal pattern profile analysis. Pediatr Radiol 2003; 33: 173–176.
Di Cicco M, Padoan R, Felisati G et al: Otorhinolaringologic manifestation of Smith–Magenis syndrome. Int J Pediatr Otorhinolaryngol 2001; 59: 147–150.
Liburd N, Ghosh M, Riazuddin S et al: Novel mutations of MYO15A associated with profound deafness in consanguineous families and moderately severe hearing loss in a patient with Smith–Magenis syndrome. Hum Genet 2001; 109: 535–541.
Finucane BM, Jaeger ER, Kurtz MB, Weinstein M, Scott Jr CI : Eye abnormalities in the Smith–Magenis contiguous gene deletion syndrome. Am J Med Genet 1993; 45: 443–446.
Gropman AL, Duncan WC, Smith AC : Neurologic and developmental features of the Smith–Magenis syndrome (del 17p11.2). Pediatr Neurol 2006; 34: 337–350.
Smith AC, Dykens E, Greenberg F : Behavioral phenotype of Smith–Magenis syndrome (del 17p11.2). Am J Med Genet 1998; 81: 179–185.
Girirajan S, Mendoza-Londono R, Vlangos CN et al: Smith–Magenis syndrome and moyamoya disease in a patient with del(17)(p11.2p13.1). Am J Med Genet A 2007; 143: 999–1008.
Smith AC, Dykens E, Greenberg F : Sleep disturbance in Smith–Magenis syndrome (del 17 p11.2). Am J Med Genet 1998; 81: 186–191.
De Leersnyder H, De Blois MC, Claustrat B et al: Inversion of the circadian rhythm of melatonin in the Smith–Magenis syndrome. J Pediatr 2001; 139: 111–116.
Potocki L, Glaze D, Tan DX et al: Circadian rhythm abnormalities of melatonin in Smith–Magenis syndrome. J Med Genet 2000; 37: 428–433.
Dykens EM, Finucane BM, Gayley C : Brief report: cognitive and behavioral profiles in persons with Smith–Magenis syndrome. J Autism Dev Disord 1997; 27: 203–211.
Finucane BM, Konar D, Haas-Givler B, Kurtz MB, Scott Jr CI : The spasmodic upper-body squeeze: a characteristic behavior in Smith–Magenis syndrome. Dev Med Child Neurol 1994; 36: 78–83.
Natacci F, Corrado L, Pierri M et al: Patient with large 17p11.2 deletion presenting with Smith–Magenis syndrome and Joubert syndrome phenotype. Am J Med Genet 2000; 95: 467–472.
Boddaert N, De Leersnyder H, Bourgeois M, Munnich A, Brunelle F, Zilbovicius M : Anatomical and functional brain imaging evidence of lenticulo-insular anomalies in Smith–Magenis syndrome. Neuroimage 2004; 21: 1021–1025.
Smith AC, Gropman AL, Bailey-Wilson JE et al: Hypercholesterolemia in children with Smith–Magenis syndrome: del (17) (p11.2p11.2). Genet Med 2002; 4: 118–125.
Vlangos CN, Wilson M, Blancato J, Smith AC, Elsea SH : Diagnostic FISH probes for del(17)(p11.2p11.2) associated with Smith–Magenis syndrome should contain the RAI1 gene. Am J Med Genet A 2005; 132: 278–282.
Truong HT, Kohal SS, Baker KR et al: Diagnosing Smith-Magenis syndrome and Duplication 17p11.2 syndrome by RAI1 gene copy-number variation using Quantitative Real-time PCR. Genet Test, 2008, in press.
Girirajan S, Elsas II LJ, Devriendt K, Elsea SH : RAI1 variations in Smith–Magenis syndrome patients without 17p11.2 deletions. J Med Genet 2005; 42: 820–828.
Bi W, Saifi GM, Shaw CJ et al: Mutations of RAI1, a PHD-containing protein, in nondeletion patients with Smith–Magenis syndrome. Hum Genet 2004; 115: 515–524.
Bi W, Saifi GM, Girirajan S et al: RAI1 point mutations, CAG repeat variation, and SNP analysis in non-deletion Smith–Magenis syndrome. Am J Med Genet A 2006; 140: 2454–2463.
Lee JA, Lupski JR : Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron 2006; 52: 103–121.
Shaw CJ, Bi W, Lupski JR : Genetic proof of unequal meiotic crossovers in reciprocal deletion and duplication of 17p11.2. Am J Hum Genet 2002; 71: 1072–1081.
Shaw CJ, Lupski JR : Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet 2005; 116: 1–7.
Shaw CJ, Withers MA, Lupski JR : Uncommon deletions of the Smith–Magenis syndrome region can be recurrent when alternate low-copy repeats act as homologous recombination substrates. Am J Hum Genet 2004; 75: 75–81.
Juyal RC, Figuera LE, Hauge X et al: Molecular analyses of 17p11.2 deletions in 62 Smith–Magenis syndrome patients. Am J Hum Genet 1996; 58: 998–1007.
Lucas RE, Vlangos CN, Das P, Patel PI, Elsea SH : Genomic organisation of the approximately 1.5 Mb Smith–Magenis syndrome critical interval: transcription map, genomic contig, and candidate gene analysis. Eur J Hum Genet 2001; 9: 892–902.
Toulouse A, Rochefort D, Roussel J, Joober R, Rouleau GA : Molecular cloning and characterization of human RAI1, a gene associated with schizophrenia. Genomics 2003; 82: 162–171.
Maier D, Stumm G, Kuhn K, Preiss A : Hairless, a Drosophila gene involved in neural development, encodes a novel, serine rich protein. Mech Dev 1992; 38: 143–156.
Onodera O, Oyake M, Takano H, Ikeuchi T, Igarashi S, Tsuji S : Molecular cloning of a full-length cDNA for dentatorubral-pallidoluysian atrophy and regional expressions of the expanded alleles in the CNS. Am J Hum Genet 1995; 57: 1050–1060.
Gerber HP, Seipel K, Georgiev O et al: Transcriptional activation modulated by homopolymeric glutamine and proline stretches. Science 1994; 263: 808–811.
Aasland R, Gibson TJ, Stewart AF : The PHD finger: implications for chromatin-mediated transcriptional regulation. Trends Biochem Sci 1995; 20: 56–59.
Rekdal C, Sjottem E, Johansen T : The nuclear factor SPBP contains different functional domains and stimulates the activity of various transcriptional activators. J Biol Chem 2000; 275: 40288–40300.
Seranski P, Hoff C, Radelof U et al: RAI1 is a novel polyglutamine encoding gene that is deleted in Smith–Magenis syndrome patients. Gene 2001; 270: 69–76.
De Leersnyder H, de Blois MC, Vekemans M et al: beta(1)-adrenergic antagonists improve sleep and behavioural disturbances in a circadian disorder, Smith–Magenis syndrome. J Med Genet 2001; 38: 586–590.
De Leersnyder H, de Blois MC, Bresson JL, Sidi D, Claustrat B, Munnich A : [Inversion of the circadian melatonin rhythm in Smith–Magenis syndrome]. Rev Neurol (Paris) 2003; 159: 6S21–6S26.
Bi W, Ohyama T, Nakamura H et al: Inactivation of Rai1 in mice recapitulates phenotypes observed in chromosome engineered mouse models for Smith–Magenis syndrome. Hum Mol Genet 2005; 14: 983–995.
Acknowledgements
We thank Dr Helga Toriello and Dr Arti Pandya for critical review of the manuscript. Dr Sarah H Elsea was supported, in part, by NIH R01 HD38534 and by resources from Virginia Commonwealth University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Elsea, S., Girirajan, S. Smith–Magenis syndrome. Eur J Hum Genet 16, 412–421 (2008). https://doi.org/10.1038/sj.ejhg.5202009
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5202009
Keywords
This article is cited by
-
Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation (ROHHAD): a collaborative review of the current understanding
Clinical Autonomic Research (2023)
-
Copy number variations of chromosome 17p11.2 region in children with development delay and in fetuses with abnormal imaging findings
BMC Medical Genomics (2021)
-
Methylphenidate for attention-deficit/hyperactivity disorder in patients with Smith–Magenis syndrome: protocol for a series of N-of-1 trials
Orphanet Journal of Rare Diseases (2021)
-
Microfibril-associated glycoprotein 4 (Mfap4) regulates haematopoiesis in zebrafish
Scientific Reports (2020)
-
Rhythms of life: circadian disruption and brain disorders across the lifespan
Nature Reviews Neuroscience (2019)
