
Abstract
Mutations in PAX9 have been described for families in which inherited oligodontia characteristically involves permanent molars. Our study analysed one large family with dominantly inherited oligodontia clinically and genetically. In addition to permanent molars, some teeth were congenitally missing in the premolar, canine, and incisor regions. Measurements of tooth size revealed the reduced size of the proband's and his father's deciduous and permanent teeth. This phenotype is distinct from oligodontia phenotypes associated with mutations in PAX9. Sequencing of the PAX9 gene revealed a missense mutation in the beginning of the paired domain of the molecule, an arginine-to-tryptophan amino-acid change occurring in a position absolutely conserved in all sequenced paired box genes. A mutation of the homologous arginine of PAX6 has been shown to affect the target DNA specificity of PAX6. We suggest that a similar mechanism explains these distinct oligodontia phenotypes.
Similar content being viewed by others
Introduction
The development of human dentition is a complex process regulated by a sequence of reciprocal interactions between the epithelial and mesenchymal tissue components. Signal molecules and growth factors from several different families and their specific receptors, as well as several transcription factors have been associated with epithelial–mesenchymal signalling during tooth morphogenesis.1,2 This complex regulation is reflected in numerous inherited conditions with dental aberrations.
Agenesis of some teeth is a common human anomaly. Approximately 21% of the population lacks one or more third molars and if third molars are not considered, 8% lacks one or more teeth.3 Oligodontia, a severe and rare form of tooth agenesis, is defined as the absence of six or more permanent teeth, with the absence of less than six teeth referred to as hypodontia. For oligodontia, a prevalence of 0.16% has been reported.4 It is associated with several syndromes, but is also inherited as an isolated trait.
This far, familial nonsyndromic oligodontia is known to be involved in several mutations in two transcription factor coding genes, MSX1 and PAX9.5,6,7,8,9,10,11 Mutations in MSX1 segregate with tooth agenesis that involves predominantly second premolars and third molars. Mutations in PAX9 have been identified as the defects responsible for missing permanent molars.
PAX9 plays important roles in development and organogenesis.12,13 Pax9 knockout mice lack pharyngeal pouch derivatives, have craniofacial and limb anomalies, and fail to form teeth beyond the bud stages.12 Like the Pax9 knockouts, Msx1 knockouts also lack teeth, and their development is arrested at the bud stage.14 In contrast to mutations in man, heterozygous Pax9 and Msx1 mouse mutants exhibit no obvious defects. The lower gene dosage appears to be critical for proper development of the more complex human dentition. Furthermore, it remains unknown as to why PAX9 and MSX1 mutations appear to cause different phenotypes. It is possible that PAX9 plays a special role in the development of molar teeth, which show a development distinct from that of other permanent teeth, as they lack deciduous predecessors, instead arising directly from the distal extension of the dental lamina.
We investigated a Finnish family affected with oligodontia, in which mutational analysis of PAX9 by exon sequencing identified a novel mutation, C76T. The resulting phenotype is distinct from previously reported oligodontia phenotypes caused by mutations in PAX9.
Patients and methods
The proband visited an orthodontist at the age of six for anterior crossbite. Radiographic examination revealed multiple missing permanent teeth. His parents, paternal grandfather, and the grandfather's siblings and their offspring were then examined (Figure 1). The diagnosis of oligodontia was based on clinical examination, panoramic or periapical radiographs, and interviews.

Pedigree (circles indicate females, squares indicate males) showing affection status: closed, affected; open, unaffected. The arrow indicates the proband. Pedigree displays an autosomal-dominant mode of inheritance.
Tooth size was measured from plaster models available from the proband and his affected father and healthy mother. In more distant affected relatives, prosthetic structures and extracted teeth made the measurement of tooth size impossible. The maximal mesiodistal and buccolingual widths of teeth were measured with sliding calipers according to the method of Alvesalo.15 Measurements of permanent teeth were compared to the means and standard deviations (SD) for Finns.15 Measurements of deciduous teeth were compared to those of healthy Finnish children.16
DNA was isolated from venous blood samples by the PuregeneTM DNA purification system (Gentra, Minneapolis, MN, USA). For sequencing, exons 1–4 of PAX9 were amplified by PCR with DyNAzymeTM EXT DNA polymerase (Finnzymes, Espoo, Finland). Information on the PCR primers and PCR conditions is given in Table 1. PCR products were purified by 2% agarose gel electrophoresis and NucleoSpin® Extract 2 in 1 kit (Macherey-Nagel, Düren, Germany). Both strands of the PCR products were sequenced with the BigDye terminator chemistry and analysed on an ABI 377XXL DNA sequencer (Applied Biosystems, Foster City, CA, USA). Sequencing results were compared by BLAST2 (http://ncbi.nlm.nih.com/gorf/bl2.html) with EMBL entries AJ238381, AJ238382, and AJ238383.
For restriction-enzyme analysis, forward and reverse primers for the 5′-terminal amplicon of exon 2 were used to amplify the region spanning the mutation. The 680 bp amplified fragment was then digested with HpaII (Sigma, St Louis, MO, USA), and the digestion products were run in a 3% agarose gel.
The study was approved by the Ethics Committee of the Institute of Dentistry, and consent was obtained from family members.
Results
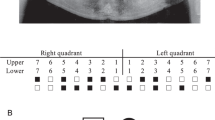
The medical history of family members revealed no health problems or disorders related to hair, nails, or sweat glands. Congenital lack of several permanent teeth, oligodontia, segregated as an autosomal-dominant trait (Figure 1). The proband had complete primary dentition, but the radiograph revealed several missing permanent teeth (Figure 2). His mother had perfect permanent dentition, but in his father 14 permanent teeth were congenitally missing. The proband's grandfather (II:4) knew that he had had several congenitally missing teeth. The same was true for his brother (II:2), whose two children (III:3, III:5) presented with oligodontia. A common feature of the permanent dentition of the four affected individuals whose phenotype could be properly determined was the lack of second and third molars in both the maxilla and mandible. Missing second premolars could be verified in three affected family members. Three individuals also lacked one or more maxillary first molars. One or two lower incisors were missing in two affected males. In addition, two affected family members also lacked one or both upper permanent canines. Both maxillary lateral incisors were missing in two individuals, and in one they were peg-shaped (Table 2).

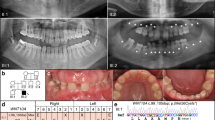
(a) Panoramic radiograph of the proband (IV:1) at age 6 years, 11 months. Stars indicate congenitally missing teeth. (b) Clinical photograph of the proband at the same age. Note the tapering crown of the permanent upper central incisors.
Tooth size was measured for the proband, his affected father, and healthy mother; the mother's tooth size exceeded that of controls. Mesiodistally, the size of the proband's deciduous teeth were of the order of −0.19 to −2.12 SD. Buccolingual dimensions were also smaller than in controls, varying from –0.19 to –2.82 SD. The most deviant teeth were the upper second primary molars, central primary incisors, and lower primary canines. We could measure only four permanent teeth that were also smaller in size. The size of the affected father's permanent teeth was also considerably reduced: mesiodistally, of the order of −0.45 to −4.18 SD and buccolingually of −0.88 to −3.46 SD smaller than in controls. The size of incisors in both jaws deviated most from Finnish norms. Measurements for central incisors and first molars of the proband and his father are presented in Table 3.
Sequencing of the candidate gene PAX9 revealed a nucleotide transition C76T in the proband and his father (Figure 3). The single base change removed an HpaII restriction-enzyme site, and this was used to test the rest of the family (Figure 4). All affected family members were heterozygous for the change, which was absent from unaffected subjects. Further tests on 100 unrelated controls were negative for this change. The C76T transition results in the substitution of a neutral tryptophan for a positively charged arginine in residue 26 of PAX9.

Detection of PAX9 mutation. (a) PAX9 gene and protein structure. Grey boxes indicate exons and darker boxes indicate the coding sequence. The black box is the paired box. All reported PAX9 mutations are depicted. (b) Chromatogram from sequencing of exon 2 of the proband (IV:1), arrow indicating the mutation. (c) Comparison of nucleotide and amino-acid sequences of the wild-type and mutant genes and proteins. HpaII recognition site is underlined.

(a) HpaII restriction-enzyme analysis of PCR products of exon 2 of PAX9. Sizes of ds-DNA molecular size markers are indicated on the right. (b) Restriction map. PCR products were 680 bp long. Restriction sites are indicated by arrows. Digestion fragment sizes in bp are indicated below the map. The mutant allele was not cleaved at the site shown (*), whereas the wild type was cleaved normally. Upon digestion, the sequence carrying the mutation resulted in an undigested fragment of 307 bp.
Discussion
For this family, in which oligodontia was inherited as an autosomal-dominant trait, our results strongly suggest that the phenotype was caused by the mutation C76T. The mutation results in the substitution of tryptophan for an arginine in exon 2 of PAX9, representing the first missense mutation this far. This mutation appeared in all affected family members, whereas all unaffected family members as well as the 100 controls were negative for this change. Sequence comparison of all available PAX sequences from different species shows that the arginine at a homologous position was absolutely conserved, whereas the neighbouring amino acids showed considerable variation (Figure 5). In humans, three mutations in homologous arginine have been reported. The arginine is mutated to leucine in PAX3 in one family with Waardenburg's syndrome type 1,17 in PAX6 to glycine in a family with anterior segment abnormalities of the eye,18 and in PAX8 to histidine in one patient with hypoplasia of the thyroid gland.19

Multiple sequence alignment of PAX sequences sorted first by species and then by homology. Only partial N-terminal sequences of the paired box are shown. Sequences were obtained from the SwissProt database and aligned with Clustal W v 1.82. (*) Conservation of amino acids among all species listed; (↑) Conserved Arg26.
This far, four different PAX9 mutations have been reported in families affected with severe lack of permanent teeth, in particular of permanent molars and premolars. Characteristically, when a premolar or molar tooth is missing, all the posterior teeth on the same hemiarch are also absent.6,8,9,11 Oligodontia of molar teeth was most pronounced in a family with complete deletion of one copy of the PAX9 gene.11 Our clinical analysis revealed that the pattern of tooth agenesis in the family we describe deviates from PAX9 phenotypes previously reported (Table 4). Several second premolars were missing, even though the neighbouring first molar existed, rather resembling the phenotype seen previously in association with mutations of MSX1. In addition to missing teeth, we noticed smaller tooth size in affected family members. Reduced permanent tooth size is common in patients with hypodontia or oligodontia.20,21 It is worth noting that in our proband, aged six, the deciduous teeth were also reduced in size. Our results indicate that reduction of tooth size associated with tooth agenesis may be caused by a mutation of a single gene.
Nine different genes code for the human PAX molecules, all containing a paired box domain characteristic of all PAX proteins.13,22 For PAX6, the crystallographic structure of the paired domain has been resolved.23 Sequence comparison shows strong homology, with a 75% amino-acid identity in the paired box between PAX9 and PAX6. In PAX6, homologous Arg26 is localized in the first α-helix of the N-terminal subdomain of the paired domain and is one of the residues that directly contacts DNA.23 A mutation of this arginine is reported in a family with various anterior segment malformations of the eye.18 As mutations in the human PAX6 typically produce aniridia, the relatively milder phenotypes of the reported R26G mutation suggest that it creates a hypomorphic allele.24 The R26G mutant of PAX6 fails to bind a subset of paired domain-binding sites, while retaining the ability to bind the rest of the sites and to transactivate promoters containing those sites.25 Differential DNA-binding capacity has also been shown for PAX5 experimentally mutated for homologous arginine.26 These effects have been explained by the bipartite structure of the paired domain and the binding sites.26 It is possible that mutation of R26 of PAX9 also affects the transcription of target genes differentially. It has been demonstrated that the mesenchymal expression of Msx1 is substantially downregulated in Pax9 (−/−) mouse embryos.12 It is possible that the effect of this mutation is analogous to that of the PAX6 hypomorphic mutation and may cause a selective reduction in PAX9 binding to sites that regulate MSX1 expression. This may explain why the phenotype in the family reported here is distinct from that of previously reported families.
In summary, we describe a three-generation family with dominantly inherited oligodontia that affects all tooth groups, as well as tooth size in both deciduous and permanent teeth. The condition most probably results from a mutation in the PAX9 gene. It seems that in addition to molars, normal development of all teeth, including incisors, canines, and premolars, depends on the function of the PAX9 gene.
References
Jernvall J, Thesleff I : Reiterative signaling and patterning during mammalian tooth morphogenesis. Mech Dev 2000; 92: 19–29.
Nieminen P, Pekkanen M, Åberg T, Thesleff I : Gene Expression in Tooth. Developmental Biology Programme Finland: Institute of Biotechnology, University of Helsinki, 2003, http://biteit.helsinki.fi.
Haavikko K : Hypodontia of permanent teeth. Proc Finn Dent Soc 1971; 67: 219–225.
Rolling S, Poulsen S : Oligodontia in Danish schoolchildren. Acta Odontol Scand 2001; 59: 111–112.
Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE : A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet 1996; 13: 417–421.
Stockton DW, Das P, Goldenberg M, D'Souza RN, Patel PI : Mutation of PAX9 is associated with oligodontia. Nat Genet 2000; 24: 18–19.
van den Boogaard MJH, Dorland M, Beemer FA, van Amstel HKP : MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet 2000; 24: 342–343.
Nieminen P, Arte S, Tanner D et al: Identification of a nonsense mutation in the PAX9 gene in molar oligodontia. Eur J Hum Genet 2001; 9: 743–746.
Frazier-Bowers SA, Guo DC, Cavender A et al: A novel mutation in human PAX9 causes molar oligodontia. J Dent Res 2002; 81: 129–133.
Lidral AC, Reising BC : The role of MSX1 in human tooth agenesis. J Dent Res 2002; 81: 274–278.
Das P, Stockton DW, Bauer C et al: Haploinsufficiency of PAX9 is associated with autosomal dominant hypodontia. Hum Genet 2002; 110: 371–376.
Peters H, Neubuser A, Kratochwil K, Balling R : Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev 1998; 12: 2735–2747.
Dahl E, Koseki H, Balling R : Pax genes and organogenesis. BioEssays 1997; 19: 755–765.
Satokata I, Maas R : Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 1994; 6: 348–356.
Alvesalo L : The influence of sex-chromosome genes on tooth size in man; Dissertation Institute of Dentistry, University of Oulu, 1970.
Kotilainen J : Development of dentition in conditions with multiple congenital anomalies; Dissertation Institute of Dentistry, University of Helsinki, 1996.
Hoth CF, Milunsky A, Lipsky N, Sheffer R, Clarren SK, Baldwin CT : Mutations in the paired domain of the human PAX3 gene cause Klein–Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I). Am J Hum Genet 1993; 52: 455–462.
Hanson IM, Fletcher JM, Jordan T et al: Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters' anomaly. Nat Genet 1994; 6: 168–173.
Macchia PE, Lapi P, Krude H et al: PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet 1998; 19: 83–86.
Schalk-van der Weide, Steen WH, Beemer FA, Bosman F : Reductions in size and left–right asymmetry of teeth in human oligodontia. Arch Oral Biol 1994; 39: 935–939.
Brook AH, Elcock C, al-Sharood MH, McKeown HF, Khalaf K, Smith RN : Further studies of a model for the etiology of anomalies of tooth number and size in humans. Connect Tissue Res 2002; 43: 289–295.
Chi N, Epstein JA : Getting your Pax straight: Pax proteins in development and disease. Trends Genet 2002; 18: 41–47.
Xu HE, Rould MA, Xu W, Epstein JA, Maas RL, Pabo CO : Crystal structure of the human Pax6 paired domain–DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev 1999; 13: 1263–1275.
Hanson I, van Heyningen V : Pax6: more than meets the eye. Trends Genet 1995; 11: 268–272.
Tang HK, Chao LY, Saunders GF : Functional analysis of paired box missense mutations in the PAX6 gene. Hum Mol Genet 1997; 6: 381–386.
Czerny T, Schaffner G, Busslinger M : DNA sequence recognition by Pax proteins: bipartite structure of the paired domain and its binding site. Genes Dev 1993; 7: 2048–2061.
Acknowledgements
We thank all the members of the affected family for their participation. We express our gratitude to Marjatta Kivekäs and Anneli Sinkkonen for their help in the laboratory. This study was financially supported by the Finnish Dental Society.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lammi, L., Halonen, K., Pirinen, S. et al. A missense mutation in PAX9 in a family with distinct phenotype of oligodontia. Eur J Hum Genet 11, 866–871 (2003). https://doi.org/10.1038/sj.ejhg.5201060
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201060
Keywords
This article is cited by
-
Development of a novel ex vivo organ culture system to improve preservation methods of regenerative tissues
Scientific Reports (2023)
-
Nonsyndromic oligodontia
Journal of Orofacial Orthopedics / Fortschritte der Kieferorthopädie (2017)
-
Management of a child with severe hypodontia in the mixed dentition stage of development
European Archives of Paediatric Dentistry (2014)
-
Genetic background of nonsyndromic oligodontia: a systematic review and meta-analysis
Journal of Orofacial Orthopedics / Fortschritte der Kieferorthopädie (2013)
-
Radiographic study of tooth agenesis in the Turkish population
Oral Radiology (2010)
