
Abstract
This review describes the changes of spinal neuronal function that occur after a motor complete spinal cord injury (cSCI) in humans. In healthy subjects, polysynaptic spinal reflex (SR) evoked by non-noxious tibial nerve stimulation consists of an early SR component and rarely a late SR component. Soon after a cSCI, SR and locomotor activity are absent. After spinal shock; however, an early SR component re-appears associated with the recovery of locomotor activity in response to appropriate peripheral afferent input. Clinical signs of spasticity take place in the following months, largely as a result of non-neuronal changes. After around 1 year, the locomotor and SR activity undergo fundamental changes, that is, the electromyographic amplitude in the leg muscles during assisted locomotion exhaust rapidly, accompanied by a shift from early to dominant late SR components. The exhaustion of locomotor activity is also observed in non-ambulatory patients with an incomplete spinal cord injury (SCI). At about 1 year after injury, in most cSCI subjects the neuronal dysfunction is fully established and remains more or less stable in the following years. It is assumed that in chronic SCI, the patient's immobility resulting in a reduced input from supraspinal and peripheral sources leads to a predominance of inhibitory drive within spinal neuronal circuitries underlying locomotor pattern and SR generation. Training of spinal interneuronal circuits including the enhancement of an appropriate afferent input might serve as an intervention to prevent neuronal dysfunction after an SCI.
Similar content being viewed by others

Introduction
There are several promising neuroregenerative and neuroprotective treatments that are directed to limit neuronal damage and/or induce neuronal regeneration after a spinal cord injury (SCI)1, 2, 3 and they will enter the clinic within the next decade.4, 5, 6 However, the auspicious results in animal experiments often cannot be replicated in humans. For example, the application of regeneration-facilitating olfactory ensheathing cells led to a recovery of function after an SCI in rodents,7, 8, 9, 10 but had no or only a poor effect on the neurological deficit in humans with SCI11, 12, 13, 14, 15 in well-controlled14 but also rather uncontrolled studies.11, 12, 13, 15
The main problem is the time of the treatment onset. It is known that early treatment onset may help to prevent scar formation16 and therefore, a part of regeneration could occur before scar formation is established. Another important aspect concerns the intrinsic capacity of central neurons to regenerate which leads to the necessity to treat as early as possible after an SCI. Therefore, the treatments in rat models are usually administered soon after the injury.17, 18, 19, 20 In humans, treatment after the SCI is often delayed because at a later stage the clinical condition is more stable. A preservation of the function of spinal neuronal circuits below the level of lesion, however, is an important prerequisite for the success of any kind of regeneration-inducing therapy.5
Studies with chronic complete spinal cord injury (cSCI) subjects during the past few years provided some evidence that the function of spinal neuronal circuits below the level of the lesion is impaired.21, 22 The purpose of this review is to depict the behavior of spinal neuronal circuits deprived from appropriate supraspinal and afferent input and to discuss potential countermeasures to prevent neuronal dysfunction below the level of lesion in the chronic stage of SCI.
Neuronal dysfunction after SCI
As spinal shock resolves after an acute SCI, the reappearance of polysynaptic spinal reflex (SR) function is associated with the recovery of locomotor EMG activity in rats,23 and people with cSCI through assisted leg movements.21, 24, 25 In humans, during the subsequent weeks, the amplitude of locomotor EMG signals and SR activity increases. However, compared with recordings made in healthy individuals, the amplitude of leg muscle EMG activity evoked during assisted locomotion with body-weight support is much reduced.26, 27
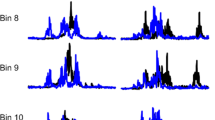
The neuronal activities that underlie locomotion and SR reach a steady state some months after a human SCI.18, 28 A cSCI at this stage is characterized by the development of spasticity with exaggerated tendon tap reflexes, increased muscle tone and spasms. Electrophysiological recordings of polysynaptic SR show successively smaller amplitudes of the early SR component and increasing amplitudes of the late SR component, while H-reflexes are unchanged.28, 29 At a chronic stage of cSCI only late SR components are observed (see Figure 1). Secondarily occurring non-neuronal changes, that is, alteration in muscle mechanic, rather than a neuronal hyperactivity, are assumed to be responsible to increased muscle tone at the subacute stage after the SCI.30, 31, 32 In addition, the difference in the level of muscle activity between active (movement) and passive (clinical) conditions is lesser in spastic limbs than in unaffected limbs.33

SR responses in healthy and chronic cSCI subjects. Representative examples of SR responses in the ipsilateral tibialis anterior muscle evoked by tibial nerve stimulation at the medial malleolus in a healthy (age 25 years) and a chronic (4.3 years since injury) cSCI (age 23 years) subject. At the onset of the electromyographic recordings, stimulus artifacts are present.
Both spinal and supraspinal lesions lead to both loss of supraspinal drive and defective use of afferent input. These changes obviously affect the behavior of short and long-latency SR and lead to paresis and maladaptation of the movement pattern. Changes in the mechanical properties of muscle fibers34 result in spastic muscle tone, which partly compensates for the paresis.35 In patients with incomplete SCI, these non-neuronal changes allow to keep posture during walking, as seen in the spastic movement disorder.
In humans, characteristic changes in neuronal behavior occur approximately 1 year after a severe SCI: after some minutes of assisted locomotion, the EMG amplitude decreases nearly to a noise level.22 This phenomenon called ‘EMG exhaustion’ cannot be reversed by locomotor training and is more pronounced in the leg flexor than in the extensor muscles. So far, no condition in animal models is known that corresponds to the phenomenon of EMG exhaustion in human SCI. The exhaustion of locomotor activity is assumed to take place at a premotoneuronal (that is, spinal interneuronal) level.36 Two observations support this assumption: first, the muscle action potentials and H-reflexes do not change in amplitude during repetitive nerve stimulation22, 37 and second, EMG activity of all leg muscles became enhanced during spasms induced by stumbling, despite of an exhaustion of locomotor activity at the end of a training session.22
Corresponding to the EMG exhaustion phenomenon during assisted walking, SR behavior undergoes changes too insofar that a second late SR component can be evoked by tibial nerve stimulation, while the amplitude of the early component decreases.21 There obviously exists a temporal relationship between the decrease of the early SR component, the increase of the late SR component and the degree of exhaustion of locomotor activity (see Figure 2).21

Time course of SRs and locomotor activity. Representative examples of SR behavior and locomotor activity during assisted locomotion at (a) early, (b) transition and (c) chronic stages after a complete SCI. SRs see Figure 1. Locomotor activity of four leg muscles is shown at the beginning of assisted locomotion (left side) and 10 min thereafter (right side). From Dietz et al.21
A partial loss of EMG activity occurs already from the beginning of a training session and encloses a rarefaction of EMG activity/potentials, while during EMG exhaustion the EMG activity is present at the beginning of assisted locomotion and fades out over 5–10 min. This partial loss of EMG activity that is observed mainly in the leg flexors seems to occur independently of the exhaustion of EMG activity.21 Transsynaptic degeneration of motoneurons might be the cause of this loss of EMG activity as suggested by rodent studies.38 Neuronal circuits in the spinal cord of rats strongly depend on the input that is lost after an SCI.39, 40 Also in human SCI there is indirect evidence suggesting a transsynaptic degeneration of spinal neurons after an SCI.41, 42, 43, 44 For example, using the threshold tracking technique, excitability of peroneal nerve axons was found to drop down distal to the site of injury in SCI subjects.43
The EMG exhaustion phenomenon is observed in the presence of long-lasting immobility, regardless of the completeness of the SCI. Although most SCI subjects classified according to the American Spinal Injury Association Impairment Scale (AIS) as AIS A and B show the exhaustion phenomenon,21 patients with sensorimotor incomplete SCI (AIS C and D) who regularly perform stepping movements show no EMG exhaustion and their early SR component remains dominant. In contrast, incomplete SCI subjects who are wheelchair-bound show the same exhaustion of EMG activity as do AIS A SCI patients, and a late SR component is dominant.21
Mechanisms of neuronal dysfunction
So far, the cause of the EMG exhaustion phenomenon remains unclear. Our currently favored assumption is that a loss of supraspinal and appropriate peripheral input causes a predominance of inhibitory influences on the locomotor pattern, leading to the decrease of EMG amplitude during assisted locomotion.36 In vertebrates, there exists a close interaction of excitatory and inhibitory interneuronal activity within the pattern-generating neuronal circuits shaping the locomotor pattern.45 After an SCI, the excitatory neuronal circuits become deprived of appropriate afferent input and concurrently more-centrally controlled inhibitory neuronal circuits might become predominant, resulting in a weakening of the function of excitatory neuronal circuits. These changes might be reflected in the facilitation of long-latency pathways that mediate inhibitory signals, resulting in the inhibition of the early—and thereby mediating the late—SR component. Indeed, such basic changes in the balance between excitatory and inhibitory inputs to spinal neuronal circuits have been described in cats with SCI46, 47 in which walking function could markedly be improved by blocking of inhibitory transmission.48
The two following observations support the evidence that changes in locomotor and SR function in chronic SCI are caused by a shift towards the predominance of inhibitory neuronal circuits and not necessarily to a degradation of spinal neuronal function: first, the intact locomotor networks observed even more than 25 years after an SCI, and second, the modifiability of EMG exhaustion (and SR changes) in incomplete SCI subjects by functional training.
To what extent an improvement in locomotor function by training is associated with a shift to a dominant early SR component is yet unclear. In addition, it would be of interest to see whether the SR component pattern can serve as a marker to identify preserved locomotor function, that is, daily walking ability in patients suffering an SCI.
Potential countermeasures
The preservation of neuronal function below the level of lesion is of crucial importance for the success of future regeneration-inducing treatments after a severe SCI. Presently, regeneration-inducing therapies should not be performed in chronic SCI subjects showing a neuronal dysfunction until appropriate countermeasures are developed. The weak effect of a non-functional recovery described recently for single chronic subjects after olfactory ensheathing cell transplantation15 would even be compatible with the neuronal dysfunction described here. In the future, an appropriate timing of combined regeneration- and plasticity-enhancing therapies should be taken into account.49, 50 Functional training seems to have an important role in the prevention of neuronal dysfunction. However, the exhaustion phenomenon could not yet be reversed by a locomotor training in the chronic stage of a motor complete SCI.22 Preliminary observations indicate that the neuronal dysfunction is partially reversible in non-ambulatory AIS C subjects by locomotor training combined with functional electrical stimulation (unpublished observation). In fact, an improvement of ambulatory function through intensive locomotor training can be achieved in chronic incomplete SCI subjects.51 By such a functional training therapy inhibitory signals might become blocked as demonstrated in the cat.48
Actually, several questions have to be answered. How soon after an SCI should locomotor training be started in order to prevent neuronal dysfunction? Which qualitative features does the training require? Would the provision of some artificial afferent input, for example, repetitive electrical stimulation of flexor-reflex afferents and/or cutaneous afferents, be sufficient to prevent neuronal dysfunction? The aim has to be to induce regeneration before a neuronal dysfunction is established.
Conclusions
A dysfunction of spinal neuronal circuits, underlying locomotion and associated reflex, develops after a severe SCI. The dysfunction of spinal neurons chronically deprived of supraspinal and appropriate peripheral input is reflected in an exhaustion of locomotor EMG activity with an associated shift from dominant early to dominant late SR components. These changes are suggested to be due to an imbalance of excitatory and inhibitory neuronal circuits resulting in the emergence of a bias to inhibitory signals within spinal neuronal circuitries. Further research is required to more precisely define the nature of the neuronal dysfunction, that is, the changes of excitatory and inhibitory activity of neuronal circuits below the level of lesion. In addition, an animal model of chronic SCI might be able to elucidate the neuronal mechanisms underlying the neuronal dysfunction after an SCI and facilitates the development of appropriate countermeasures.
References
Raineteau O, Schwab ME . Plasticity of motor systems after incomplete spinal cord injury. Nat Rev Neurosci 2001; 2: 263–273.
Buchli AD, Rouiller E, Mueller R, Dietz V, Schwab ME . Repair of the injured spinal cord. A joint approach of basic and clinical research. Neuro-Degenerative Diseases 2007; 4: 51–56.
Zeman RJ, Feng Y, Peng H, Visintainer PF, Moorthy CR, Couldwell WT et al. X-irradiation of the contusion site improves locomotor and histological outcomes in spinal cord-injured rats. Exp neurol 2001; 172: 228–234.
Raisman G . A promising therapeutic approach to spinal cord repair. J R So Med 2003; 96: 259–261.
Dietz V, Curt A . Neurological aspects of spinal-cord repair: promises and challenges. Lancet neurol 2006; 5: 688–694.
Deumens R, Koopmans GC, Joosten EA . Regeneration of descending axon tracts after spinal cord injury. Prog Neurobiol 2005; 77: 57–89.
Richter MW, Roskams AJ . Olfactory ensheathing cell transplantation following spinal cord injury: hype or hope? Exp neurol 2008; 209: 353–367.
Ramon-Cueto A, Cordero MI, Santos-Benito FF, Avila J . Functional recovery of paraplegic rats and motor axon regeneration in their spinal cords by olfactory ensheathing glia. Neuron 2000; 25: 425–435.
Li Y, Field PM, Raisman G . Repair of adult rat corticospinal tract by transplants of olfactory ensheathing cells. Science (New York, N.Y) 1997; 277: 2000–2002.
Barnett SC, Chang L . Olfactory ensheathing cells and CNS repair: going solo or in need of a friend? Trends Neurosci 2004; 27: 54–60.
Chhabra HS, Lima C, Sachdeva S, Mittal A, Nigam V, Chaturvedi D et al. Autologous olfactory mucosal transplant in chronic spinal cord injury: an Indian Pilot Study. Spinal Cord 2009; 47: 887–895.
Curt A, Dietz V . Controversial treatments for spinal-cord injuries. Lancet 2005; 365: 841.
Dobkin BH, Curt A, Guest J . Cellular transplants in China: observational study from the largest human experiment in chronic spinal cord injury. Neurorehabil Neural Repair 2006; 20: 5–13.
Mackay-Sim A, Feron F, Cochrane J, Bassingthwaighte L, Bayliss C, Davies W et al. Autologous olfactory ensheathing cell transplantation in human paraplegia: a 3-year clinical trial. Brain 2008; 131 (Part 9): 2376–2386.
Lima C, Escada P, Pratas-Vital J, Branco C, Arcangeli CA, Lazzeri G et al. Olfactory mucosal autografts and rehabilitation for chronic traumatic spinal cord injury. Neurorehabil Neural Repair 2010; 24: 10–22.
Klapka N, Hermanns S, Straten G, Masanneck C, Duis S, Hamers FP et al. Suppression of fibrous scarring in spinal cord injury of rat promotes long-distance regeneration of corticospinal tract axons, rescue of primary motoneurons in somatosensory cortex and significant functional recovery. Eur J Neurosci 2005; 22: 3047–3058.
Houle JD, Tessler A . Repair of chronic spinal cord injury. Exp Neurol 2003; 182: 247–260.
Karimi-Abdolrezaee S, Eftekharpour E, Wang J, Morshead CM, Fehlings MG . Delayed transplantation of adult neural precursor cells promotes remyelination and functional neurological recovery after spinal cord injury. J Neurosci 2006; 26: 3377–3389.
Nomura H, Baladie B, Katayama Y, Morshead CM, Shoichet MS, Tator CH . Delayed implantation of intramedullary chitosan channels containing nerve grafts promotes extensive axonal regeneration after spinal cord injury. Neurosurgery 2008; 63: 127–141;discussion 141–3.
Ye JH, Houle JD . Treatment of the chronically injured spinal cord with neurotrophic factors can promote axonal regeneration from supraspinal neurons. Exp Neurol 1997; 143: 70–81.
Dietz V, Grillner S, Trepp A, Hubli M, Bolliger M . Changes in spinal reflex and locomotor activity after a complete spinal cord injury: a common mechanism? Brain 2009; 132 (Part 8): 2196–2205.
Dietz V, Muller R . Degradation of neuronal function following a spinal cord injury: mechanisms and countermeasures. Brain 2004; 127 (Part 10): 2221–2231.
Lavrov I, Gerasimenko YP, Ichiyama RM, Courtine G, Zhong H, Roy RR et al. Plasticity of spinal cord reflexes after a complete transection in adult rats: relationship to stepping ability. J Neurophysiol 2006; 96: 1699–1710.
Dietz V, Colombo G, Jensen L . Locomotor activity in spinal man. Lancet 1994; 344: 1260–1263.
Dietz V, Muller R, Colombo G . Locomotor activity in spinal man: significance of afferent input from joint and load receptors. Brain 2002; 125 (Part 12): 2626–2634.
Halder P, Curt A, Brem S, Lang-Dullenkopf A, Bucher K, Kollias S et al. Preserved aspects of cortical foot control in paraplegia. NeuroImage 2006; 31: 692–698.
Dietz V, Harkema SJ . Locomotor activity in spinal cord-injured persons. J Appl Physiol 2004; 96: 1954–1960.
Hiersemenzel LP, Curt A, Dietz V . From spinal shock to spasticity: neuronal adaptations to a spinal cord injury. Neurology 2000; 54: 1574–1582.
Jones CA, Yang JF . Reflex behavior during walking in incomplete spinal-cord-injured subjects. Exp Neurol 1994; 128: 239–248.
Dietz V, Colombo G, Jensen L, Baumgartner L . Locomotor capacity of spinal cord in paraplegic patients. Ann Neurol 1995; 37: 574–582.
Dietz V, Quintern J, Berger W . Electrophysiological studies of gait in spasticity and rigidity. Evidence that altered mechanical properties of muscle contribute to hypertonia. Brain 1981; 104: 431–449.
O’Dwyer NJ, Ada L, Neilson PD . Spasticity and muscle contracture following stroke. Brain 1996; 119 (Part 5): 1737–1749.
Ibrahim IK, Berger W, Trippel M, Dietz V . Stretch-induced electromyographic activity and torque in spastic elbow muscles. Differential modulation of reflex activity in passive and active motor tasks. Brain 1993; 116 (Part 4): 971–989.
Lieber RL, Friden J . Spasticity causes a fundamental rearrangement of muscle-joint interaction. Muscle Nerve 2002; 25: 265–270.
Dietz V, Sinkjaer T . Spastic movement disorder: impaired reflex function and altered muscle mechanics. Lancet Neurol 2007; 6: 725–733.
Dietz V . Behavior of spinal neurons deprived of supraspinal input. Nat Rev Neurol 2010; 6: 167–174.
Muller R, Dietz V . Neuronal function in chronic spinal cord injury: divergence between locomotor and flexion- and H-reflex activity. Clin Neurophysiol 2006; 117: 1499–1507.
Burns AS, Lemay MA, Tessler A . Abnormal spontaneous potentials in distal muscles in animal models of spinal cord injury. Muscle Nerve 2005; 31: 46–51.
Ginsberg SD, Martin LJ . Axonal transection in adult rat brain induces transsynaptic apoptosis and persistent atrophy of target neurons. J Neurotrauma 2002; 19: 99–109.
Wu YP, Ling EA . Transsynaptic changes of neurons and associated microglial reaction in the spinal cord of rats following middle cerebral artery occlusion. Neurosci Lett 1998; 256: 41–44.
Aisen ML, Brown W, Rubin M . Electrophysiologic changes in lumbar spinal cord after cervical cord injury. Neurology 1992; 42 (3 Part 1): 623–626.
Chang CW . Evident transsynaptic degeneration of motor neurons after spinal cord injury: a study of neuromuscular jitter by axonal microstimulation. Am J Phy Med Rehabil/Association of Academic Physiatrists 1998; 77: 118–121.
Lin CS, Macefield VG, Elam M, Wallin BG, Engel S, Kiernan MC . Axonal changes in spinal cord injured patients distal to the site of injury. Brain 2007; 130 (Part 4): 985–994.
Nyboer VJ, Johnson HE . Electromyographic findings in lower extremities of patients with traumatic quadriplegia. Arch Phys Med Rehabil 1971; 52: 256–259.
Grillner S, Deliagina T, Ekeberg O, el Manira A, Hill RH, Lansner A et al. Neural networks that co-ordinate locomotion and body orientation in lamprey. Trends Neurosci 1995; 18: 270–279.
Ichiyama RM, Broman J, Edgerton VR, Havton LA . Ultrastructural synaptic features differ between alpha- and gamma-motoneurons innervating the tibialis anterior muscle in the rat. J Comp Neurol 2006; 499: 306–315.
Tillakaratne NJ, de Leon RD, Hoang TX, Roy RR, Edgerton VR, Tobin AJ . Use-dependent modulation of inhibitory capacity in the feline lumbar spinal cord. J Neurosci 2002; 22: 3130–3143.
de Leon RD, Tamaki H, Hodgson JA, Roy RR, Edgerton VR . Hindlimb locomotor and postural training modulates glycinergic inhibition in the spinal cord of the adult spinal cat. J Neurophysiol 1999; 82: 359–369.
Maier IC, Ichiyama RM, Courtine G, Schnell L, Lavrov I, Edgerton VR et al. Differential effects of anti-Nogo-A antibody treatment and treadmill training in rats with incomplete spinal cord injury. Brain 2009; 132 (Part 6): 1426–1440.
Garcia-Alias G, Barkhuysen S, Buckle M, Fawcett JW . Chondroitinase ABC treatment opens a window of opportunity for task-specific rehabilitation. Nat Neurosci 2009; 12: 1145–1151.
Wirz M, Zemon DH, Rupp R, Scheel A, Colombo G, Dietz V et al. Effectiveness of automated locomotor training in patients with chronic incomplete spinal cord injury: a multicenter trial. Arch Phys Med Rehabil 2005; 86: 672–680.
Acknowledgements
This work was supported by the Seventh Framework Program of the European Commission (project ‘Spinal Cord Repair’; HEALTH-F2–2007–201144).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Hubli, M., Bolliger, M. & Dietz, V. Neuronal dysfunction in chronic spinal cord injury. Spinal Cord 49, 582–587 (2011). https://doi.org/10.1038/sc.2010.147
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sc.2010.147
Keywords
This article is cited by
-
Abnormal cutaneous flexor reflex activity during controlled isometric plantarflexion in human spinal cord injury spasticity syndrome
Spinal Cord (2016)
-
Strategies and lessons in spinal cord injury rehabilitation
Current Physical Medicine and Rehabilitation Reports (2015)
-
Neuromuscular training based on whole body vibration in children with spina bifida: a retrospective analysis of a new physiotherapy treatment program
Child's Nervous System (2015)
-
The physiological basis of neurorehabilitation - locomotor training after spinal cord injury
Journal of NeuroEngineering and Rehabilitation (2013)
