
Abstract
We present a table-top beamline providing a soft X-ray supercontinuum extending up to 370 eV from high-order harmonic generation with sub-13 fs 1300 nm driving pulses and simultaneous production of sub-5 fs pulses centered at 800 nm. Optimization of high harmonic generation in a long and dense gas medium yields a photon flux of ~ 1.4 × 106 photons/s/1% bandwidth at 300 eV. The temporal resolution of X-ray transient absorption experiments with this beamline is measured to be 11 fs for 800 nm excitation. This dual-wavelength approach, combined with high flux and high spectral and temporal resolution soft X-ray absorption spectroscopy, is a new route to the study of ultrafast electronic dynamics in carbon-containing molecules and materials at the carbon K-edge.
Similar content being viewed by others
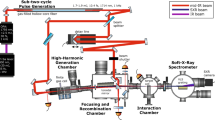

Introduction
Transient absorption spectroscopy with high-order harmonic generation (HHG) sources is now established as a powerful technique to probe ultrafast dynamics in atoms1,2,3, molecules4,5,6 and solids7,8,9,10 (see e.g. ref. 11 for a review). Being an all-optical method, in which the timing is done first and the spectral resolution separately, the time and energy resolution are independent and not subject to an uncertainty relationship. It can therefore combine the attosecond temporal capabilities of HHG sources with the atomic-specificity and the chemical environment sensitivity of X-ray spectroscopy. In X-ray absorption spectroscopy, intense absorption features are located around the so-called atomic edges. Many of the heavier elements show spectroscopic signatures in the extreme ultra-violet region (XUV, 10–100 eV) corresponding to L2,3, M4,5 or N4,5 edges. However, several key elements are not accessible in this spectral domain, including sulfur (L2,3 -edge at 163 eV), carbon (K-edge at 284 eV), nitrogen (K-edge at 410 eV) and oxygen (K-edge at 540 eV), that are present in the majority of organic and biochemically-relevant molecules and materials. Recent experimental efforts (see e.g. ref. 12 for a review) have thus focused on increasing the photon energy available from table-top X-ray sources, to reach the soft X-ray domain (SXR, 100–1000 eV). This is achieved by taking advantage of the scaling of the cutoff energy in the HHG process with intensity I and wavelength λ of the driving laser field as Ecutoff ∝ Iλ2 (ref. 13). While the first SXR photons produced with a HHG source utilized high intensity 800 nm driving wavelength in a strong ionization regime14,15, the strategy that is now preferred is to use longer wavelength drivers in the short-wave infrared (SWIR)12,16,17,18 and mid-infrared19.
In order to provide a spectral supercontinuum instead of discrete harmonic peaks, as well as allow the production of isolated attosecond pulses and therefore considerably improve the temporal resolution, the use of few-cycle SWIR pulses to drive SXR HHG has recently been developed. Indeed, the 9 fs output of an optical parametric chirped pulse amplifier centered at 1600 nm generated a SXR continuum up to 350 eV in neon20. By taking advantage of the polarization gating technique with 12 fs, 1800 nm pulses, and the appropriate choice of metallic filters to compensate for the SXR dispersion21, an isolated pulse of 53 attoseconds extending above 300 eV was measured22. Likewise, more conventional approaches relying on the post-compression of 30–50 fs idler pulses from Ti:sapphire-pumped Optical Parametric Amplifiers (OPA) produced SXR radiation supporting isolated attosecond pulses23,24. All of these studies use few-cycle SWIR pulses in the 1600–1850 nm range, which offer the advantages of (i) passive Carrier-Envelope Phase (CEP) stabilization25 and (ii) the use of transmission through bulk glass for post-compression26. However, compared to shorter wavelengths, they suffer unfavourable scaling of the HHG efficiency with the driving wavelength, ∝ λ−6 at the single-atom level27,28. In those conditions, a single absorption spectrum requires accumulation of the signal over a few minutes22,29. Reduced integration times will be decisive for the application of SXR transient absorption spectroscopy to the study of attosecond chemical dynamics in systems containing S, C or N atoms30,31,32. Therefore, a trade-off comprising a continuous spectrum extending above the carbon K-edge with high flux and few-femtosecond temporal resolution would be desirable. Moreover, transient absorption in the XUV typically employs a fraction of energy of the few-cycle visible-near IR pulses used for HHG as a pump via strong-field4,6 or single-photon excitation5,8,9,10. Due to the smaller photon energy in the SWIR, using a similar approach for SXR transient absorption considerably reduces the pumping excitation capabilities. The availability of an intense, ultrashort, second color pulse in the visible-near infrared synchronous with the SXR-generating SWIR used as a pump would circumvent this issue. Note however that field-driven carrier motion in a semi-metal33 and tunnel ionization of a low ionization potential molecule34 with λ > 1600 nm few-cycle pulses have recently been observed.
In this work, a table-top beamline simultaneously producing few-cycle SWIR and vis-NIR pulses to be used in ultrafast pump-probe experiments in the SXR is presented. The two-cycle pulses centered at 1300 nm are used to drive HHG in helium, providing a continuous spectrum extending up to 370 eV, with ~1.4 × 106 photons/s/1% bandwidth at 300 eV. We show that the SXR pulses produced enable the measurement of high resolution absorption spectra in the 170–300 eV range within a few seconds. When used in combination with sub-5 fs vis-NIR pulses, the temporal resolution achieved in a transient absorption experiment at 250 eV is ~11 fs.
Description of the table-top dual-wavelength beamline
The beamline is summarized in Fig. 1. It is based on a 13 mJ, 800 nm, 30 fs, 1 kHz Ti:Sapphire laser (Coherent Legend Elite Duo), whose energy is split into 11 mJ and 2 mJ to produce the probe and pump pulses, respectively, used in time-resolved X-ray transient absorption experiments. This section details the simultaneous compression of SWIR (centered at 1300 nm or 1400 nm) and visible-near infrared (centered at 800 nm) pulses, and the characteristics of the soft X-ray spectrometer.

Summary of the beamline. Soft X-ray pulses are produced from high-order harmonic generation with few-cycle SWIR pulses in helium. Few-cycle pulses centered at 800 nm are simultaneously produced and are used as a pump in ultrafast transient absorption experiments. The different steps are detailed in the text. A typical X-ray camera image is shown at the bottom right. The drop in signal after ~280 eV is due to carbon contamination of the X-ray optics, causing absorption at the carbon K-edge.
Production of few-cycle short-wave infrared pulses
Few-cycle pulses centered at 1300 nm in the SWIR are produced using an OPA followed by spectral broadening in a hollow-core fiber filled with a rare gas35 and compression with chirped mirrors36. The 11 mJ 800 nm beam is further split into 0.5 mJ and 10.5 mJ for use in a two-stage OPA, which converts 800 nm to SWIR (1300 nm). The two-stage system is obtained from Light Conversion and is designed to provide CEP stability of the signal pulses. The first stage is a low energy OPA (TOPAS Prime), pumped by 0.5 mJ, which provides 90 μJ of 2 μm pulses in the idler. Due to the parametric amplification process, the idler pulses are passively CEP stabilized, with a stability of <200 mrad. They are then used as the seed for the white light generation (WLG) in a second, high energy, OPA (HE-TOPAS) pumped with the remaining 10.5 mJ of 800 nm light. This design should ensure the CEP stability of the signal pulses over the 1200–1600 nm tunable range of the OPA, regardless of the CEP stability of the pump laser. However, the following results are obtained with a CEP-averaged signal pulse.
The 2.8 mJ, 1300 nm, 40 fs output is focused with a 2-m focusing mirror to a focal size of 390 μm (at 1/e2) at the entrance of a 3-m-long, 700-μm-inner diameter stretched hollow-core fiber (HCF) filled with 0.4 bar of argon (Few-Cycle Inc.). The ratio of beam waist to fiber diameter was experimentally adjusted for best transmission through the fiber, and it was found that this slightly smaller value (0.56) compared to the optimal value for coupling to the LP01 mode37 (0.64) yielded higher output power. After broadening through self-phase modulation, the spectrum extends from 1000 nm to 1500 nm (see the dashed line in Fig. 2d), supporting 9.3 fs pulses with up to 1.45 mJ per pulse. After recollimation, the pulses are compressed with a set of four ultra-broadband dispersive mirrors (PC1816, Ultrafast Innovations GmbH) that provide an average of −150 fs2 per reflection over the 1000–1700 nm spectral range. For fine-tuning of the group-delay dispersion (GDD), the beam goes through a pair of counter-rotating 2-mm NaCl windows, which have a GDD of +47.8 fs2/mm at 1300 nm. The pulses are characterized using a home-built dispersion-free second harmonic generation Frequency-Resolved Optical Gating (SHG-FROG), whose results are shown in Fig. 2a–d. The retrieved pulse has a full width at half maximum (fwhm) in intensity of 12.8 fs, which corresponds to less than 3 optical cycles, with 80% of the energy in the main pulse. Further compression could be achieved with a better control over third-order dispersion.

Characterization of the compressed SWIR pulses produced with the OPA central wavelength set at 1300 nm (a–d) and 1400 nm (e–h) with SHG-FROG. (a,e) Measured spectrogram. (b,f) Retrieved spectrogram. (c,g) Retrieved temporal intensity (pink) and phase (grey). (d,h) Retrieved spectral intensity (pink) and phase (grey). The dashed line is the spectrum at the output of the fiber as measured with a spectrometer (Ocean Optics FLAME). Phases are shown for intensities >5% of the maximum intensity.
The ultra-broadband dispersive mirrors are optimized for chirp compensation over a 700-nm bandwidth in the SWIR. In order to test the tunability of the HCF compressor, the central wavelength of the OPA is increased to 1400 nm, the rest of the system remaining identical. The spectrum is broadened in 0.6 bar of argon and extends from 1050 to 1600 nm (Fig. 2h), with up to 1.15 mJ per pulse. From the SHG-FROG measurement and retrieval shown in Fig. 2e–g, the pulses are compressed down to 9.6 fs fwhm, that is 2 cycles at 1400 nm, with 88% of the energy in the main pulse. Thus, we see that the system allows wavelength tuning in the SWIR over a 100 nm range.
Simultaneous production of few-cycle visible-near IR pulses
The 2 mJ pulses at 800 nm split before the pumping of the OPA stages (see Fig. 1) are compressed to few-cycle duration using the same method35 in a second, 400-μm-inner diameter stretched HCF filled with 1.5 bar of neon. After propagation over two meters to compensate for the delay acquired by the SWIR pulse in the OPA stages, the beam is focused with a 2-m focusing lens to a 280 μm spot size at the entrance to the fiber. At the output, the pulse energy is 0.8 mJ with a spectrum covering 550–950 nm (Fig. 3c). Compression is achieved with reflections on sixteen ultra-broadband dispersive mirrors (PC1332, Ultrafast Innovations GmbH), transmission through fused silica wedges and finally through a 2-mm-thick Ammonium Dihydrogen Phosphate (ADP) crystal, which corrects for third-order dispersion38. The visible-near IR (vis-NIR) pulses are characterized with a d-scan module (Sphere Ultrafast Photonics)39, whose results are shown in Fig. 3. The d-scan phase retrieval algorithm indicates a pulse duration of 4.75 fs fwhm, which is 1.1 times the Fourier limit, with 75% of the intensity in the main pulse. Compression of the vis-NIR pulses to 1.8 cycle is realized at the same time as the compression of the SWIR pulses described in the previous section. Therefore, these two pulses can be used together in a pump-probe experiment. We note that shorter pulse durations in the vis-NIR with similar pulse energy can be achieved with this method38, and could most likely be obtained here if the compression of the Ti:Sa laser could be tuned independently for pumping the OPA stages and the vis-NIR HCF compressor.

Characterization of the few-cycle 800 nm pulses used as a pump. (a) Measured dscan. (b) Retrieved dscan. (c) Measured spectrum (red) and retrieved spectral phase (grey) of the compressed pulses. (d) Retrieved temporal intensity (red) and Fourier-limited pulse (black).
Soft X-ray spectrometer
The soft X-ray (SXR) pulses are produced via high-order harmonic generation by focusing 0.7 mJ of the few-cycle SWIR pulses with a f = 30 cm lens down to a 80 μm (at 1/e2) diameter focus (~2 × 1015 W/cm2) into a 4-mm-long gas cell continuously flowing with helium at a backing pressure of 2.8 bar (see Fig. 1). The use of a lens is unusual with few-cycle driving pulses5,22– 24,40; however, the near 0 GDD of fused silica at 1300 nm (−2.4 fs2/mm) provides minimal pulse lengthening from chirp and geometrical sources41. The gas cell is surrounded by a 7 cm edge-length cube for differential pumping, sealed along the propagation axis with cellophane tape through which holes are laser-produced. The cube is evacuated by a roots blower with 900 m3/h pumping speed, such that the pressure inside is calculated to be ~1 Torr and is measured to be ~1 × 10−3 Torr in the surrounding chamber. After HHG, the SWIR light is filtered out with a metallic foil (Zr, Sn, Ag, Ti or Al can be used depending on the spectral region of interest). The SXR is focused by a Ni-coated toroidal mirror at 4° grazing angle into the sample, which can be a gas cell or a solid membrane. Changes between different samples can be easily achieved with motorized stages in the two dimensions perpendicular to the propagation axis. The spectrum is dispersed by an aberration-corrected concave grating and imaged onto an X-ray CCD camera (Greateyes, GE 2048 512 BI UV1). The camera is movable along the grating focal plane to fully capture the SXR spectrum. Depending on the spectral region of interest, two different gratings can be used, optimized for 5–25 nm (50–247 eV, Hitachi 001-0660) or 1–6 nm (207–1239 eV, Hitachi 001–0659), referred to as the low- and high-energy gratings in the following. The grating rotation and camera position can be adjusted to optimize the spectral resolution. The resolution of the spectrometer has been determined using the Ar 2p\({}_{3/2}^{-1}\)4s absorption line42 to be 325 and 190 meV fwhm at 244.39 eV, corresponding to ΔE/E = 1.3 × 10−3 and ΔE/E = 7.8 × 10−4 for the two gratings, respectively. While the resolution of the high-energy grating is higher, its increased groove density results in a lower efficiency by a factor of ~3. Unless otherwise mentioned, all the spectra shown in the following are measured using the low-energy grating.
Optimization of the photon flux at the carbon K-edge
Driving wavelength
Here we study how the high-harmonic flux in the cutoff region differ for two few-cycle driving pulses of different wavelengths in the SWIR. Indeed, at the single-atom level, the HHG efficiency scales with λ−6 (refs. 27,28). However, this goes together with an increase in the cutoff photon energy as λ2 (ref. 13), and calculations have shown that the atomic response can be macroscopically overcome under certain phase-matching conditions17. In the previous section, the tunability of the few-cycle SWIR source was demonstrated. We therefore compare the SXR spectra produced when driving the HHG in the 4 mm gas cell with the 1300 nm and 1400 nm pulses characterized in Fig. 2. The HHG spectra produced in these conditions are shown in Fig. 4a. From the measured spectra, the photon fluxes are calculated at the source using the known quantum efficiency of the CCD camera and number of electrons generated per X-ray photon of a given energy, as well as tabulated toroidal mirror reflectivity, metallic filter transmission, and grating efficiency. At both wavelengths, the spectra are continuous and extend from below 150 eV (not shown in Fig. 4 because of the limited size of the camera chip) to above 300 eV. As expected, the maximum photon energy produced with few-cycle pulses centered at 1400 nm is higher compared to 1300 nm. The photon flux is 2 to 10 times larger at the 1300 nm driving wavelength over the 150–280 eV spectral range. However, in the carbon K-edge region above 280 eV, the two drivers produce similar high harmonic fluxes. The measured photon fluxes, in photons/s in 1% and 10% bandwidth at 300 eV, are summarized in Table 1 and compared to literature values for SXR HHG sources driven by few-cycle SWIR pulses.

Optimization of the flux at 300 eV with the driving wavelength and the gas cell length. (a) Soft X-ray high-order harmonic spectra generated in helium with few-cycles pulses with the OPA central wavelength set at 1300 nm (red, solid) and 1400 nm (green, dashed) in a 4 mm gas cell. (b) Soft X-ray high-order harmonic spectra generated in helium with few-cycles pulses with the OPA central wavelength set at 1300 nm in 1 mm (blue, dotted), 4 mm (red, solid) and 7.5 cm (yellow, dashed) length gas cells. Further optimization of the SWIR HCF gas pressure for HHG with the 7.5 cm cell yields the optimal solid yellow curve.
HHG gas cell length
The question of macroscopic effects in HHG with SWIR pulses has been the subject of recent experimental and theoretical work16,17,18,23. Here we compare the SXR flux obtained in helium with few-cycle 1300 nm pulses in continuously flowed gas cells of different lengths. The spectrum obtained in a 4 mm gas cell described previously is reproduced in red, solid in Fig. 4b. Johnson et al. showed that, in the so-called overdriven regime, plasma defocusing effects strongly reshape the HHG driving pulse, already in the tails of the pressure distribution around the gas cell24. In order to limit these effects, a shorter gas cell of 1 mm length (1 mm outer diameter and 500 μm inner diameter) is used in place of the 4 mm one described before. The corresponding HHG spectrum, generated in 4 bar of helium, is shown in blue on Fig. 4b. No increase of the cutoff is observed, and the flux at 300 eV is 2x less than with a longer interaction medium (Table 1).
Since the absorption length at 300 eV of 2 bar of helium is ~1 cm, a longer HHG medium was also tried43. SXR pulses are generated in a 7.5 cm long tube, sealed at the entrance with a 500 μm thick window and with a teflon foil through which a hole is produced with the laser at the end. The focus of the SWIR is towards the exit of the cell, therefore being qualified as semi-infinite34,44. No nonlinear effects in the entrance window were observed at the working intensities. A reduced ~2 bar helium pressure is required to optimize HHG in the semi-infinite gas cell, so differential pumping is not necessary. Instead, the roots blower pump is used to back the turbomolecular pump attached to the HHG chamber, where the pressure is measured to be ~10−3 Torr in these conditions. As shown in dashed yellow in Fig. 4b, this gas cell produces an order of magnitude more photon flux at 300 eV, with the maximum achievable photon energy remaining identical. Therefore, the semi-infinite gas cell is used in the following experiments. Following this observation, the other experimental parameters were optimized to maximize the SXR flux. Broadening the SWIR pulse in 0.7 bar of argon, instead of 0.4 bar previously, produces the solid yellow spectrum visible on Fig. 4b, with 1.4 × 106 photons/s in 1% bandwidth at 300 eV (see Table 1). The results call for further investigations of the role of ionization in HHG with few-cycle pulses in a semi-infinite gas cell.
We note that continuous SXR spectra are obtained in all the reported cases, but this observation is not proof of the production of isolated attosecond pulses - these spectra are measured with random CEP and the lack of CEP stability can cause random phase shifts in the HHG spectra that then appear as a continuum45. The semi-infinite gas cell yields more photon flux around the carbon K-edge, but is probably less suited for the production of isolated attosecond pulses. However, few-fs SXR pulses at the carbon K-edge (combined with a few-fs pump) would be able to probe ultrafast dynamics on the few tens of femtoseconds timescale, such as passage through conical intersections in organic molecules. Temporal characterization of the SXR pulses22,46 is needed, in particular in the long gas cell case, but this is out of the scope of the current paper. However, the temporal resolution of the beamline is estimated from a transient absorption experiment in the following section. It is limited by the durations of the vis-NIR pulse (see Fig. 3), the SXR probe, and the optical path fluctuation of the ~15 m long interferometer. The delay jitter has been measured using a balanced optical cross-correlator47 to be 2 fs rms over 10 s.
Absorption spectroscopy in the soft X-ray
Measurement of absorption spectra with the high-harmonic source
In order to demonstrate the suitability of the SXR HHG source, X-ray absorption spectra are typically measured22,24,40,48,49,50,51. As seen on Fig. 1, a gas cell can be inserted in the SXR beam path at the focus of the toroidal mirror. This allows the measurement of spectra such as shown in Fig. 5, where the semi-infinite gas cell and 1300 nm pulses are used for HHG (dashed yellow spectrum on Fig. 4b). The absorbance, in optical densities (OD), is defined as \(A=-\,{{\rm{\log }}}_{10}\frac{I}{{I}_{0}}\), where I and I0 are the background-corrected spectra measured with and without the sample, respectively. The high flux of our beamline and the continuous, broad SXR spectrum allow the measurement of absorption spectra in a few seconds only, without scanning the photon energy. The absorption spectrum of SF6 around the sulfur L2,3 and L1 edges displayed in Fig. 5a is acquired in 6 seconds (3 seconds with and without the sample). The spin-orbit split doublet corresponding to the excitation of a S 2p electron to the unoccupied a1g molecular orbital is well-resolved, as well as a series of excitations to molecular Rydberg states around 180 eV52. Above the S 2p−1 ionization threshold, excitation to quasi-bound states t2g and eg is visible, as well as the weaker transition of the S 2s electron to a t1u molecular orbital around 240 eV. The short acquisition times and the high spectral resolution should allow the observation of ultrafast dynamics in the SF6 molecule53. Similarly, absorption spectra of argon around the L2,3 edge around 250 eV are obtained in 6 seconds (Fig. 5b). At the spectral resolution of 325 meV, three peaks in the series converging to the Ar 2p−1 threshold can be resolved29,42. Despite the lower flux at higher photon energies (see Fig. 4 and Table 1), the absorption spectrum of CO2 around the carbon K-edge can also be measured, this time in 20 seconds (10 seconds with and without the sample). Three main features of the spectrum can be identified, including the main C 1s to π* transition54 (Fig. 5c).

X-ray near-edge absorption spectra measured with the SXR HHG source in (a) SF6, (b) Ar and (c) CO2.
Ultrafast transient absorption in argon near the L2,3 edge
In order to realize pump-probe experiments, the sub-5 fs vis-NIR pulse is time-delayed and focused in the sample gas cell with a concave mirror and recombined with the SXR on a hole mirror (see Fig. 1). The optical light is filtered by a metallic foil before the SXR spectrometer. A representative transient absorption experiment is performed in argon, using SXR produced with the 7.5 cm long HHG gas cell filled with 2 bar of helium. The delay-dependent absorption spectra are shown in Fig. 6a. At each delay point τ, the absorbance A is calculated as \(A(\tau )=-\,{{\rm{\log }}}_{10}\frac{I(\tau )}{{I}_{0}(\tau )}\), where the reference spectrum I0 is reconstructed by using a Fourier low-pass filter directly on the measured signal I(τ). This procedure allows for removal of the intensity fluctuations of the SXR spectra and is well-suited in the case of narrow absorption lines3. In Fig. 6a, A then represents the absorption with respect to the continuum absorption background. Here, the SXR excites 2p core-excited states of argon (Fig. 5b). In the presence of the vis-NIR pulse, with a polarization parallel to the SXR polarization and intensity of ~3 × 1013 W/cm2, the absorption lines are ac Stark-shifted. This corresponds to a decrease in absorbance at the position of the static lineshape. The spectral resolution (325 meV at this energy, see previous section) prevents the observation of hyperbolic sidebands and the direct measurement of the temporal decay of these states2,55. However, the delay dependence of the pump-induced bleach of the absorption line can give an upper bound of the temporal resolution of our experiment. The changes in absorbance as a function of the SXR-NIR delay for the 2p\({}_{1/2}^{-1}\) 3d, 2p\({}_{3/2}^{-1}\) 3d and 2p\({}_{3/2}^{-1}\) 4s states are shown in Fig. 6b–d, respectively. For these three states, the changes can be fit with a Gaussian function of ~11 fs fwhm. We note that in a similar experiment employing SXR and two-cycle 1700 nm pulses, Chew et al. observed sub-cycle oscillations that were attributed to a signature of tunnel ionization29. Due to averaging of the CEP, these oscillations are not resolved in our experiments. It would be interesting to study such processes in the future, as the calculated Keldysh parameter in our experiment is also in the tunneling regime but different than at 1700 nm.

Transient absorption spectroscopy in the vincinity of the Ar L2,3 edge with sub-5 fs vis-NIR pulses. (a) Soft X-ray absorption spectra of argon as a function of the delay between the SXR and vis-NIR pulses. Negative delay values correspond to the SXR arriving first. At each time step the spectrum is acquired during a period of 15 s, for a total measurement time of 38 min. (b–d) Lineouts of (a) at the 2p\({}_{1/2}^{-1}\) 3d energy (b), 2p\({}_{3/2}^{-1}\) 3d energy (c) and 2p\({}_{3/2}^{-1}\) 4s energy (d). The blue dots are the experimental data and the red line is a Gaussian fit, whose full width at half-maximum duration is indicated.
Discussion
The simultaneous production of millijoule-level sub-13 fs pulses at 1300 nm and sub-5 fs pulses at 800 nm with two parallel hollow-core fiber compressors has been demonstrated. The few-cycle SWIR pulses can produce a high-flux high-harmonic supercontinuum in the soft X-ray region, with ~4.5 × 107 photons/s in 1% bandwidth at 200 eV and ~1.4 × 106 photons/s in 1% bandwidth at 300 eV. This brightness allows measurement of absorption spectra in the gas phase in a few seconds, an order of magnitude improvement compared to existing SXR HHG sources based on few-cycle driving pulses at longer wavelength22,29,48. The demonstration of increased photon flux using a short SWIR wavelength and a long HHG medium represents an important step towards the observation of attosecond dynamics in solution phase with transient absorption in the water window56. The dual-wavelength approach furthermore allows the strong-field excitation of molecules4,34,38 or single-photon pumping across a majority of semiconductor bandgaps8,9, including 2D semiconductors57. The broadband, few-cycle vis-NIR pulse could additionally be upconverted to few-fs tunable UV pulses58, opening new possibilities for the study of ultrafast excited-state dynamics in molecules59,60,61. A temporal resolution of ~11 fs is measured in a pump-probe experiment. This duration is already enough to consider the observation of ultrafast chemical dynamics at the C K-edge30,31,53,59. Improved CEP stability and feedback control over the delay will allow attosecond transient absorption experiments in the SXR in the future.
References
Goulielmakis, E. et al. Real-time observation of valence electron motion. Nature 466, 739, https://doi.org/10.1038/nature09212 (2010).
Wang, H. et al. Attosecond time-resolved autoionization of argon. Physical Review Letters 105, 143002, https://doi.org/10.1103/PhysRevLett.105.143002 (2010).
Ott, C. et al. Reconstruction and control of a time-dependent two-electron wave packet. Nature 516, 374, https://doi.org/10.1038/nature14026 (2014).
Wei, Z. et al. Elucidating the origins of multimode vibrational coherences of polyatomic molecules induced by intense laser fields. Nature Communications 8, 735, https://doi.org/10.1038/s41467-017-00848-2 (2017).
Kobayashi, Y., Chang, K. F., Zeng, T., Neumark, D. M. & Leone, S. R. Direct mapping of curve-crossing dynamics in IBr by attosecond transient absorption spectroscopy. Science 365, 79–83, https://doi.org/10.1126/science.aax0076 (2019).
Timmers, H. et al. Disentangling conical intersection and coherent molecular dynamics in methyl bromide with attosecond transient absorption spectroscopy. Nature Communications 10, 3133, https://doi.org/10.1038/s41467-019-10789-7 (2019).
Schultze, M. et al. Controlling dielectrics with the electric field of light. Nature 493, 75, https://doi.org/10.1038/nature11720 (2013).
Schultze, M. et al. Attosecond band-gap dynamics in silicon. Science 346, 1348–1352, https://doi.org/10.1126/science.1260311 (2014).
Zürch, M. et al. Direct and simultaneous observation of ultrafast electron and hole dynamics in germanium. Nature Communications 8, 15734, https://doi.org/10.1038/ncomms15734 (2017).
Schlaepfer, F. et al. Attosecond optical-field-enhanced carrier injection into the GaAs conduction band. Nature Physics 14, 560 (2018).
Geneaux, R., Marroux, H. J., Guggenmos, A., Neumark, D. M. & Leone, S. R. Transient absorption spectroscopy using high harmonic generation: a review of ultrafast x-ray dynamics in molecules and solids. Philosophical Transactions of the Royal Society A 377, 20170463, https://doi.org/10.1098/rsta.2017.0463 (2019).
Ren, X. et al. Attosecond light sources in the water window. Journal of Optics 20, 023001, https://doi.org/10.1088/2040-8986/aaa394 (2018).
Krause, J. L., Schafer, K. J. & Kulander, K. C. High-order harmonic generation from atoms and ions in the high intensity regime. Physical Review Letters 68, 3535–3538, https://doi.org/10.1103/PhysRevLett.68.3535 (1992).
Spielmann, C. et al. Generation of coherent X-rays in the water window using 5-femtosecond laser pulses. Science 278, 661–664, https://doi.org/10.1126/science.278.5338.661 (1997).
Chang, Z., Rundquist, A., Wang, H., Murnane, M. M. & Kapteyn, H. C. Generation of coherent soft X rays at 2.7 nm using high harmonics. Physical Review Letters 79, 2967–2970, https://doi.org/10.1103/PhysRevLett.79.2967 (1997).
Takahashi, E. J., Kanai, T., Ishikawa, K. L., Nabekawa, Y. & Midorikawa, K. Coherent water window X ray by phase-matched high-order harmonic generation in neutral media. Physical Review Letters 101, 253901, https://doi.org/10.1103/PhysRevLett.101.253901 (2008).
Popmintchev, T. et al. Phase matching of high harmonic generation in the soft and hard x-ray regions of the spectrum. Proceedings of the National Academy of Sciences 106, 10516–10521, https://doi.org/10.1073/pnas.0903748106 (2009).
Chen, M.-C. et al. Bright, coherent, ultrafast soft X-ray harmonics spanning the water window from a tabletop light source. Physical Review Letters 105, 173901, https://doi.org/10.1103/PhysRevLett.105.173901 (2010).
Popmintchev, T. et al. Bright coherent ultrahigh harmonics in the keV x-ray regime from mid-infrared femtosecond lasers. Science 336, 1287–1291 (2012).
Ishii, N. et al. Carrier-envelope phase-dependent high harmonic generation in the water window using few-cycle infrared pulses. Nature Communications 5, 3331, https://doi.org/10.1038/ncomms4331 (2014).
López-Martens, R. et al. Amplitude and phase control of attosecond light pulses. Physical Review Letters 94, 033001, https://doi.org/10.1103/PhysRevLett.94.033001 (2005).
Li, J. et al. 53-attosecond X-ray pulses reach the carbon K-edge. Nature Communications 8, 1–5, https://doi.org/10.1038/s41467-017-00321-0 (2017).
Teichmann, S. M., Silva, F., Cousin, S. L., Hemmer, M. & Biegert, J. 0.5-keV Soft X-ray attosecond continua. Nature Communications 7, 11493, https://doi.org/10.1038/ncomms11493 (2016).
Johnson, A. S. et al. High-flux soft x-ray harmonic generation from ionization-shaped few-cycle laser pulses. Science Advances 4,https://doi.org/10.1126/sciadv.aar3761 (2018).
Baltuška, A., Fuji, T. & Kobayashi, T. Controlling the carrier-envelope phase of ultrashort light pulses with optical parametric amplifiers. Physical Review Letters 88, 133901, https://doi.org/10.1103/PhysRevLett.88.133901 (2002).
Schmidt, B. E. et al. Compression of 1.8 μm laser pulses to sub two optical cycles with bulk material. Applied Physics Letters 96, 121109, https://doi.org/10.1063/1.3359458 (2010).
Tate, J. et al. Scaling of wave-packet dynamics in an intense midinfrared field. Physical Review Letters 98, 013901, https://doi.org/10.1103/PhysRevLett.98.013901 (2007).
Shiner, A. D. et al. Wavelength scaling of high harmonic generation efficiency. Physical Review Letters 103, 073902, https://doi.org/10.1103/PhysRevLett.103.073902 (2009).
Chew, A. et al. Attosecond transient absorption spectrum of argon at the L 2,3 edge. Phys. Rev. A 97, 031407, https://doi.org/10.1103/PhysRevA.97.031407 (2018).
Northey, T. et al. Ultrafast nonadiabatic dynamics probed by nitrogen K-edge absorption spectroscopy. Physical Chemistry Chemical Physics 22, 2667–2676, https://doi.org/10.1039/C9CP03019K (2020).
Tsuru, S. et al. Time-resolved near-edge x-ray absorption fine structure of pyrazine from electronic structure and nuclear wave packet dynamics simulations. The Journal of Chemical Physics 151, 124114, https://doi.org/10.1063/1.5115154 (2019).
Hua, W., Mukamel, S. & Luo, Y. Transient x-ray absorption spectral fingerprints of the s1 dark state in uracil. The Journal of Physical Chemistry Letters, https://doi.org/10.1021/acs.jpclett.9b02692 (2019).
Buades, B. et al. Attosecond-resolved petahertz carrier motion in semi-metallic TiS2. arXiv preprint arXiv:1808.06493 (2018).
Saito, N. et al. Real-time observation of electronic, vibrational, and rotational dynamics in nitric oxide with attosecond soft x-ray pulses at 400 eV. Optica 6, 1542–1546, https://doi.org/10.1364/OPTICA.6.001542 (2019).
Nisoli, M. et al. Compression of high-energy laser pulses below 5 fs. Optics Letters 22, 522–524, https://doi.org/10.1364/OL.22.000522 (1997).
Giguère, M. et al. Pulse compression of submillijoule few-optical-cycle infrared laser pulses using chirped mirrors. Optics Letters 34, 1894–1896, https://doi.org/10.1364/OL.34.001894 (2009).
Mahieu, B. et al. Spatial quality improvement of a ti: Sapphire laser beam by modal filtering. Applied Physics B 118, 47–60, https://doi.org/10.1007/s00340-014-5953-4 (2015).
Timmers, H. et al. Generating high-contrast, near single-cycle waveforms with third-order dispersion compensation. Optics Letters 42, 811–814, https://doi.org/10.1364/OL.42.000811 (2017).
Silva, F. et al. Simultaneous compression, characterization and phase stabilization of gw-level 1.4 cycle vis-nir femtosecond pulses using a single dispersion-scan setup. Optics Express 22, 10181–10191, https://doi.org/10.1364/OE.22.010181 (2014).
Ishii, N., Kaneshima, K., Kanai, T., Watanabe, S. & Itatani, J. Generation of sub-two-cycle millijoule infrared pulses in an optical parametric chirped-pulse amplifier and their application to soft x-ray absorption spectroscopy with high-flux high harmonics. Journal of Optics 20, 014003, https://doi.org/10.1088/2040-8986/aa99ed (2018).
Bor, Z. Distortion of femtosecond laser pulses in lenses and lens systems. Journal of Modern Optics 35, 1907–1918, https://doi.org/10.1080/713822325 (1988).
Sairanen, O.-P., Kivimäki, A., Nõmmiste, E., Aksela, H. & Aksela, S. High-resolution pre-edge structure in the inner-shell ionization threshold region of rare gases Xe, Kr, and Ar. Phys. Rev. A 54, 2834–2839, https://doi.org/10.1103/PhysRevA.54.2834 (1996).
Constant, E. et al. Optimizing high harmonic generation in absorbing gases: Model and experiment. Physical Review Letters 82, 1668–1671, https://doi.org/10.1103/PhysRevLett.82.1668 (1999).
Sutherland, J. R. et al. High harmonic generation in a semi-infinite gas cell. Optics Express 12, 4430–4436, https://doi.org/10.1364/OPEX.12.004430 (2004).
Baltuška, A. et al. Attosecond control of electronic processes by intense light fields. Nature 421, 611, https://doi.org/10.1038/nature01414 (2003).
Cousin, S. L. et al. Attosecond streaking in the water window: a new regime of attosecond pulse characterization. Physical Review X 7, 041030, https://doi.org/10.1103/PhysRevX.7.041030 (2017).
Schibli, T. R. et al. Attosecond active synchronization of passively mode-locked lasers by balanced cross correlation. Optics Letters 28, 947–949, https://doi.org/10.1364/OL.28.000947 (2003).
Johnson, A. et al. Measurement of sulfur L2,3 and carbon K edge XANES in a polythiophene film using a high harmonic supercontinuum. Structural Dynamics 3, 062603, https://doi.org/10.1063/1.4964821 (2016).
Johnson, A. S. et al. Apparatus for soft x-ray table-top high harmonic generation. Review of Scientific Instruments 89, 083110, https://doi.org/10.1063/1.5041498 (2018).
Buades, B. et al. Dispersive soft x-ray absorption fine-structure spectroscopy in graphite with an attosecond pulse. Optica 5, 502–506, https://doi.org/10.1364/OPTICA.5.000502 (2018).
Popmintchev, D. et al. Near- and extended-edge x-ray-absorption fine-structure spectroscopy using ultrafast coherent high-order harmonic supercontinua. Physical Review Letters 120, 093002, https://doi.org/10.1103/PhysRevLett.120.093002 (2018).
Hudson, E. et al. High-resolution measurements of near-edge resonances in the core-level photoionization spectra of SF6. Phys. Rev. A 47, 361–373, https://doi.org/10.1103/PhysRevA.47.361 (1993).
Pertot, Y. et al. Time-resolved x-ray absorption spectroscopy with a water window high-harmonic source. Science 355, 264–267, https://doi.org/10.1126/science.aah6114 (2017).
Sham, T. K., Yang, B. X., Kirz, J. & Tse, J. S. K-edge near-edge x-ray-absorption fine structure of oxygen- and carbon-containing molecules in the gas phase. Physical Review A 40, 652–669, https://doi.org/10.1103/PhysRevA.40.652 (1989).
Beck, A. R. et al. Attosecond transient absorption probing of electronic superpositions of bound states in neon: detection of quantum beats. New Journal of Physics 16, 113016, https://doi.org/10.1088/1367-2630/16/11/113016 (2014).
Kleine, C. et al. Soft X-ray absorption spectroscopy of aqueous solutions using a table-top femtosecond soft X-ray source. The Journal of Physical Chemistry Letters 10, 52–58, https://doi.org/10.1021/acs.jpclett.8b03420 (2019).
Manzeli, S., Ovchinnikov, D., Pasquier, D., Yazyev, O. V. & Kis, A. 2D transition metal dichalcogenides. Nature Reviews Materials 2, 17033, https://doi.org/10.1038/natrevmats.2017.33 (2017).
Varillas, R. B. et al. Microjoule-level, tunable sub-10 fs UV pulses by broadband sum-frequency generation. Optics Letters 39, 3849–3852, https://doi.org/10.1364/OL.39.003849 (2014).
Attar, A. R. et al. Femtosecond x-ray spectroscopy of an electrocyclic ring-opening reaction. Science 356, 54–59, https://doi.org/10.1126/science.aaj2198 (2017).
Bhattacherjee, A. et al. Photoinduced heterocyclic ring opening of furfural: Distinct open-chain product identification by ultrafast x-ray transient absorption spectroscopy. Journal of the American Chemical Society 140, 12538–12544, https://doi.org/10.1021/jacs.8b07155 (2018).
Schnorr, K. et al. Tracing the 267 nm-induced radical formation in dimethyl disulfide using time-resolved x-ray absorption spectroscopy. The Journal of Physical Chemistry Letters 10, 1382–1387, https://doi.org/10.1021/acs.jpclett.9b00159 (2019).
Acknowledgements
We acknowledge Profs. Roger Falcone, Eric Neuscamann, Feng Wang and Norman Yao for fruitful discussions. We thank Dr. Vincent Gruson and Dr. Stefan Haessler for helpful discussions about the pulse compression, and Dr. Romain Géneaux for comments on the manuscript. This work is supported by the W.M. Keck Foundation Grant No. 042982, the National Science Foundation through NSF MRI 1624322 and matching funds from the Lawrence Berkeley National Laboratory, the College of Chemistry, the Department of Physics, and the Vice Chancellor for Research at UC Berkeley, and the US Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Dividsion under contract DE-AC02-05CH11231, FWP no. CHPHYS01. L.B. acknowledges support from the Miller Institute for Basic Research in Science at UC Berkeley.
Author information
Authors and Affiliations
Contributions
L.B. and A.D.R. contributed equally to this work. A.D.R., P.M.K., D.M.N. and S.R.L. designed the project. L.B., A.D.R. and S.G. performed the experiments. D.M.N. and S.R.L. supervised the project. L.B. and A.D.R. wrote the manuscript, with inputs from all the authors. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barreau, L., Ross, A.D., Garg, S. et al. Efficient table-top dual-wavelength beamline for ultrafast transient absorption spectroscopy in the soft X-ray region. Sci Rep 10, 5773 (2020). https://doi.org/10.1038/s41598-020-62461-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-62461-6
This article is cited by
-
Apparatus for attosecond transient-absorption spectroscopy in the water-window soft-X-ray region
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
