
Abstract
The crossed molecular beams reactions of the 1-propynyl radical (CH3CC; X2A1) with benzene (C6H6; X1A1g) and D6-benzene (C6D6; X1A1g) were conducted to explore the formation of C9H8 isomers under single-collision conditions. The underlying reaction mechanisms were unravelled through the combination of the experimental data with electronic structure and statistical RRKM calculations. These data suggest the formation of 1-phenyl-1-propyne (C6H5CCCH3) via the barrierless addition of 1-propynyl to benzene forming a low-lying doublet C9H9 intermediate that dissociates by hydrogen atom emission via a tight transition state. In accordance with our experiments, RRKM calculations predict that the thermodynamically most stable isomer – the polycyclic aromatic hydrocarbon (PAH) indene – is not formed via this reaction. With all barriers lying below the energy of the reactants, this reaction is viable in the cold interstellar medium where several methyl-substituted molecules have been detected. Its underlying mechanism therefore advances our understanding of how methyl-substituted hydrocarbons can be formed under extreme conditions such as those found in the molecular cloud TMC-1. Implications for the chemistry of the 1-propynyl radical in astrophysical environments are also discussed.
Similar content being viewed by others
Introduction
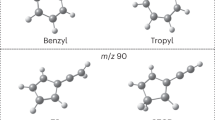
During the last decades, the C9H9 and C9H8 potential energy surfaces (PES) have received considerable attention from the astrochemistry, combustion, and chemical reaction dynamics communities in exploring the formation of indene (C9H8; [1]) along with its 1-phenyl-1-propyne (C6H5CCCH3; [2]), 1-phenyl-1,2-propadiene (C6H5CHCCH2; [3]), and 3-phenyl-1-propyne (C6H5CH2CCH; [4]) isomers in the interstellar medium (ISM) and in combustion systems (Fig. 1)1,2,3,4,5,6,7,8,9,10,11. Indene [1] represents the simplest prototype of a polycyclic aromatic hydrocarbon (PAH) containing a partially saturated pentagon fused with a benzene ring12,13,14. The carbon backbone of the indene molecule as found in corannulene (C20H10) and fullerenes like Buckminsterfullerene (C60) is central to the transition of planar PAHs like coronene (C24H12) (Fig. 2) to three-dimensional carbonaceous nanostructures and eventually soot in combustion systems and in deep space15,16,17,18. Therefore, the elementary reactions underlying the initial synthesis of PAHs carrying five-membered ring(s) is central to a complete understanding of how three-dimensional (bowl-shaped) nanostructures and ultimately soot particles are formed in extreme environments such as combustion systems and the ISM.

Energetically low-lying structural isomers of the C9H8 molecule. Enthalpies of formation (∆fH(0 K)) are given in kJ mol−1 relative to indene.

Corannulene (C20H10), Buckminsterfullerene (C60), and Coronene (C24H12).
Recent molecular beam experiments along with electronic structure calculations have been influential in exploring these early stages of chemistry via the C9H9 and C9H8 potential energy surfaces (PES). Crossed molecular beams experiments within the phenyl (C6H5)–methylacetylene (CH3CCH) system revealed the formation of 1-phenyl-1-propyne [2] at collision energies between 91–161 kJ mol−1 via short lived doublet C9H9 reaction intermediates8,9,10,11. Crossed beams studies of the phenyl (C6H5)–allene (H2CCCH2) system exposed similar dynamics via phenyl addition to a terminal carbon atom followed by hydrogen atom elimination through a tight exit transition state and 1-phenyl-1,2-propadiene [3] formation10,11. These elementary reactions must overcome entrance barriers between 6 and 26 kJ mol−1 10,11 and therefore can only operate at elevated temperatures of combustion flames and in circumstellar envelopes close to the central carbon star where temperatures up to 2,500 K reside, but not in cold molecular clouds (10 K)19. Complementary high temperature chemical microreactor studies at 600 K exploiting vacuum ultraviolet (VUV) light to photoionize the reaction products reveal that the benzyl radical (C6H5CH2) was found to react with acetylene (C2H2) yielding solely indene [1]6. The inherent entrance barrier to reaction of 51 kJ mol−1 is much higher than the barriers in the phenyl - allene and phenyl - methylacetylene systems since the benzyl radical is resonance stabilized, but the phenyl radical is not; this leads to the loss of the resonance stabilization energy of 51 kJ mol−1 upon addition of the benzyl radical to the acetylene molecule6. Contemporary crossed molecular beams experiments at lower collision energies of about 45 kJ mol−1 and high temperature chemical reactor studies at 1,200–1,500 K along with sophisticated isotopic substitution experiments provided compelling evidence that the phenyl radical reacts with allene (C3H4) and methylacetylene (C3H4) forming indene (C9H8) [1] along with non-PAH isomers 1-phenyl-1-propyne [2], 1-phenyl-1,2-propadiene [3], and possibly 3-phenyl-1-propyne [4]1,5. Generally, these studies exposed that elementary reactions of acetylene and allene/methylacetylene with aromatic radicals can access the C9H9 and C9H8 PESs. However, although the overall reactions were exoergic between 7 and 148 kJ mol−1, these studies suggest that the synthesis of any C9H8 isomer involves an entrance barrier to addition between 6 and 51 kJ mol−1 thus blocking these pathways in cold molecular clouds like Taurus (TMC-1) and Orion (OMC-1)20,21,22.
Here, we present the results of crossed molecular beams experiments of the 1-propynyl radical (CH3CC; X2A1) with benzene (C6H6; X1A1g) and D6-benzene (C6D6; X1A1g) and combine these data with novel electronic structure calculations on the C9H9 PES. These studies reveal an overall exoergic, but in contrast to previous systems, entrance-barrierless reaction accessing the C9H9 surface via addition of the 1-propynyl radical to the benzene ring leading eventually to the formation of the 1-phenyl-1-propyne product (C6H5CCCH3) along with atomic hydrogen. Our investigations propose that 1-phenyl-1-propyne could exist and be readily observable in cold molecular clouds. Methylacetylene, which has been observed toward TMC-123, is known to fragment to 1-propynyl upon interaction with ultraviolet radiation24,25,26. If formed, the 1-propynyl radical could react rapidly with benzene27 upon collision to yield 1-phenyl-1-propyne which may be susceptible to observation due to its large dipole moment of 0.48 Debye.
Experimental Results
Laboratory frame
We monitored potential products formed from the reactive scattering of the 1-propynyl radical (CH3CC; 39 amu) with benzene (C6H6; 78 amu) along ΘCM at m/z = 117 (C9H9+) and 116 (C9H8+) to assess the formation of a persistent reaction intermediate (adduct) and/or hydrogen loss reaction product, respectively. Signals were observed for each m/z value. However, the TOF spectra recorded at each m/z depict an identical pattern after scaling and thus originate from the same channel, namely the formation of C9H8 (116 amu) by elimination of atomic hydrogen (1 amu). The signal at m/z = 117 therefore arises from the natural distribution of carbon atom isotopes yielding 13CC8H8 occurring at level of about 9.9%. Following confirmation of the atomic hydrogen loss product channel, we recorded 5.6 × 106 TOF spectra of nascent C9H8 in the LAB frame at m/z = 116 in 2.5° intervals from 21.75° to 58.25° (Fig. 3). The TOF spectra were then normalized with respect to ΘCM and integrated to yield the laboratory angular distribution (Fig. 3), which is nearly symmetric around ΘCM thereby suggesting the presence of indirect reaction dynamics via one or more relatively long-lived C9H9 intermediate(s) preceding dissociation to C9H8 plus atomic hydrogen. The laboratory data were fit by forward convolution using a single reaction channel, namely CH3CC (39 amu) + C6H6 (78 amu) → C9H8 (116 amu) + H (1 amu) with a reaction cross section proportional to EC−1/3 for a barrier-less reaction within the line-of-centre model (section 5)28.

(a) Time-of-flight spectra and (b) laboratory angular distribution recorded at m/z 116 (C9H8+) for products formed in the reaction of the 1-propynyl radical with benzene. The circles represent the experimental data and the solid lines the best fits.
To trace the source of the hydrogen atom emission, we substituted the benzene reactant with D6-benzene (C6D6) and performed a second experiment with CH3CC (39 amu) plus C6D6 (84 amu) to determine if the atom is lost from the 1-propynyl or benzene reactant. We probed the atomic hydrogen and atomic deuterium loss channels along ΘCM at m/z = 122 (C9H2D6+) and 121 (C9H3D5+), respectively. A very strong signal was recorded at m/z 121 while a weaker signal was discernible at m/z = 122. By comparison with the ratios recovered in the CH3CC - C6H6 reaction for the 13CC8H8/C9H8 product(s), we conclude that the TOF spectrum at m/z = 122 owes to 13C-enrichment in the form of 13CC8H3D5 and that only the atomic deuterium loss channel to form C9H3D5 is observed. Importantly, the TOF recorded at ΘCM can be fit using the CM functions derived from the hydrogenated system, which suggests the products formed in each experiment are isotopologues (Fig. 4).

Time-of-flight (TOF) spectra for the reaction of the 1-propynyl radical with D6-benzene at m/z = 121 (C9H3D5+). The open circles represent the experimental data, and the red line represents the fit obtained from the forward-convolution routine.
Centre-of-mass frame
In the laboratory frame, it is clear that the reaction product(s) are formed via the loss of an aromatic hydrogen atom forming one or more C9H8 isomers. Details of the reaction coordinate are encoded in the product distribution and can be revealed in the centre-of-mass (CM) frame, from which we now examine via the CM translational energy P(ET) and angular T(θ) flux distributions (Fig. 5). The P(ET) terminates at 198 ± 27 kJ mol−1 and represents the maximum energy available Eavail to the reaction system. Taken together with the collision energy EC of 40.8 ± 0.5 kJ mol−1, we obtain a reaction energy for this process of ∆rG = −157 ± 27 kJ mol−1, where these quantities have been related through energy conservation via Eavail = EC − ∆rG. While the most probable ET occurs near 28 ± 4 kJ mol−1, nascent C9H8 products carry an average translational energy of 57 ± 8 kJ mol−1, suggesting that only about 29 ± 6% of Eavail is disposed into translational degrees of freedom. The high fraction of energy appearing as rovibrational excitation and nonzero peaking of the P(ET) are markers for a reaction mechanism that forms products indirectly via activated C9H9 intermediate(s) that must overcome a barrier to disscoiation28. The symmetry of the T(θ) distribution about θ = 90° along with the nonzero intensity at all angles together indicate the reaction proceeds through a long-lived C9H9 intermediate complex. Furthermore, the maximum in the distribution at θ = 90° strongly suggests the C9H9 intermediate decomposes via a transition state that emits atomic hydrogen perpendicular to its rotational plane, i.e. parallel to the total angular momentum vector as defined by the initial conditions of the experiment28,29.

The (top) center-of-mass translational energy P(ET) and (bottom) angular T(θ) flux distributions for the reaction of the 1-propynyl radical with benzene forming C9H8 isomer(s) by atomic hydrogen emission. Solid red lines represent the best fits while shaded areas indicate the experimental error limits.
Discussion
We now combine the experimental findings with electronic structure calculations to elucidate the underlying reaction dynamics and the nature of the structural isomer(s) produced by considering three aromatic C9H8 isomers. These are indene (p1), 1-phenyl-1-propyne (p2), and 1-phenyl-1,2-propadiene (p3) with computed reaction energies of −262 ± 6, −151 ± 6, and −142 ± 6 kJ mol−1, respectively. Considering the experimentally derived reaction energy of −157 ± 27 kJ mol−1, the computational data support the formation of the C9H8 isomers p2 and/or p3 via hydrogen atom loss. Continuing on the basis of energetics, indene (p1) can be reasonably excluded from consideration among the detected reaction products where the formation of the indene isomer would increase the maximum translational energy beyond the error limits (±27 kJ mol−1) of the P(ET) to about 300 kJ mol−1. By examining the reaction potential energy surface (PES), we can gain deeper insight into the underlying reaction mechanism to form C9H8 isomers via the bimolecular reaction of 1-propynyl with benzene (Fig. 6; Supplemental Information).

Schematic representation of the potential energy surface of the reaction of the 1-propynyl radical with benzene leading to the C9H8 isomers indene (p1), 1-phenyl-1-propyne (p2), and 1-phenyl-1,2-propadiene (p3). Details connecting i2 to p1 are found in ref.1.
The 1-propynyl radical can add to the π-electron system of the benzene molecule without an entrance barrier (Fig. S1) forming a C9H9 intermediate i1 featuring the propynyl substituted to the six-membered ring that lies 208 kJ mol−1 below the energy of the separated reactants. Intermediate i1 can dissociate via ts2 by eliminating the newly-formed sp3 hydrogen atom on the six-membered ring (Fig. S2) nearly perpendicular to the orbital plane of the molecule at an angle of 88.5° (Fig. 7) to form 1-phenyl-1-propyne (p2). The ts2 exit transition state is characterized by the restoration of aromaticity and hence is rather tight at 44 kJ mol−1 above the p2 product channel. Alternatively, the sp3 hydrogen atom on i1 can migrate to the C1 exocyclic position over a 148 kJ mol−1 barrier (ts1) to form i2 gaining an additional 76 kJ mol−1 in stability. From i2, each of the three C9H8 isomers p1, p2, and p3 can be accessed (Fig. S2). Elimination of the exocyclic C1 or methyl hydrogen atoms results in the formation of p2 or 1-phenyl-1,2-propadiene (p3) via ts3 or ts4, respectively. Unlike the i1 → p2 + H pathway where the hydrogen atom is ejected nearly parallel to the total angular momentum vector via ts2, the exit transition states ts3 and ts4 connecting i2 → p2 + H and i2 → p3 + H eliminate the hydrogen atom at angles of 20° and 0° with respect to the orbital plane of the molecule (Fig. 7). The minimum energy path to p1, not explicitly considered here since it was computed previously, involves extensive isomerization through an additional 8 intermediates and 9 transition states1,5.

Computed geometries of the exit transition states leading to the formation of 1-phenyl-propyne (p2) and 1-phenyl-1,2-propadiene (p3). The angle for each departing hydrogen atom is given with respect to the rotational plane of the decomposing complex.
Although both isomers p2 and p3 can account for the experimental translational energy of C9H8, a close inspection of the C9H9 PES alongside our isotopically labelled study permits a clarification of the experimental reaction dynamics. The CH3CC – C6D6 reactive scattering experiment exposed the exclusive formation of C9H3D5 via elimination of atomic deuterium originating at the benzene reactant. Isotopologues of indene p1, if formed, would give signals for atomic hydrogen (C9H2D6) and deuterium loss (C9H3D5) at a ratio of about 1:1 via a decomposing bicyclic intermediate1,5, whereas D6-p3 (C6D5CDCCH2) would form by elimination of a methyl hydrogen atom from D6-i2. Hence, only D5-p2 (C6D5CCCH3) can account for this observation via dissociation of intermediates D6-i1 or D6-i2 (Fig. 8). Recalling that the best-fit T(θ) distribution peaked sideways at 90°, we note that of the three dissociation channels considered, only the i1 → ts2 → p2 + H path supports this finding where the hydrogen emitted by ts2 occurs nearly parallel to the principal axis with the greatest moment of inertia (Ic = 1.16 × 10−44 kg m2) at an angle of 1.5°. For an asymmetric top decomposing at the top of an exit barrier, microcanonical transition state theory has shown that hydrogen atom emission from a long-lived complex along the principal axis of the greatest moment of inertia (C) results in sideways scattering30,31, as observed in this experiment (Fig. 5). The i2 → ts3 → p2 + H and i2 → ts4 → p3 + H pathways do not give rise to sideways scattering where ts4 dissociates in the AB plane and ts3 emits its hydrogen atom nearly perpendicular to its C axis. We also note the average ET for C9H8 this experiment is 57 ± 8 kJ mol−1 and is a close match to the exit barrier of 44 ± 5 kJ mol−1 for the p2 + H product channel. At the experimental collision energy of 40.8 ± 0.5 kJ mol−1 each of the p1-p3 product channels are open, however, considering the barriers to isomerization on the C9H9 PES, the low energy route to 1-phenyl-1-propyne (p2) via intermediate i1 is expected to dominate in a statistically-conforming molecular system, where the i1 → i2 barrier (ts1) is 47 kJ mol−1 greater than that required for dissociation to p2 + H via ts2. This is supported by our RRKM calculations which suggest that p2 accounts for more than 99% of the total C9H8 yield. Combined, our computational analysis and experimental data indicate the formation of 1-phenyl-1-propyne (p2) as the predominant product formed in the bimolecular reaction of 1-propynyl with benzene via a simple addition/elimination mechanism, i.e. CH3CC + C6H6 → i1 → p2 + H.

Reaction schematic for the bimolecular reaction of the 1-propynyl (CH3CC) radical with D6-benzene (C6D6) leading to C9H3D5 and C9H2D6 products via atomic deuterium and hydrogen loss pathways.
The temperature-dependent rate constant for the barrierless reaction of 1-propynyl (CH3CC) with benzene (C6H6) was evaluated using the long-range transition state theory, where the capture rate is assessed considering dipole-quadrupole, dipole-induced dipole and dispersion interactions between the reactants. The calculations gave the value of the rate constant increasing from 3.7 × 10−10 cm3 molecule−1 s−1 at 10 K to 5.4 × 10−10 and 6.5 × 10−10 cm3 molecule−1 s−1 at 100 and 300 K, respectively. These results corroborate our hypothesis that the reaction of the 1-propynyl radical with benzene should be fast even in cold molecular clouds and the rate constant can be included in astronomic kinetic models.
Lastly, we compare our results to the analogous phenyl (C6H5; X2A1) plus methylacetylene (CH3CCH; X1A1) reaction, where a hydrogen atom has been transferred from benzene to 1-propynyl – that, despite its obvious similarity with the title reaction, follows completely distinct reaction dynamics. In a low-pressure (300 Torr) high temperature (1200–1500 K) chemical reactor the phenyl reacts with methylacetylene to form the C9H8 isomers indene (p1), 1-phenyl-1-propyne (p2), 1-phenyl-1,2-propadiene (p3) at fractions of 10, 82, and 8% respectively1; in a high-energy (EC = 140 kJ mol−1) crossed molecular beams experiment, a similar outcome was observed with the formation of 1-phenyl-1-propyne (p2) via an addition/elimination reaction mechanism via a short-lived reaction adduct8. In the low-energy (EC = 45 kJ mol−1) case, however, the C6H5 plus CH3CCH reaction results in the formation of indene (p1) as confirmed through a series of isotopic experiments and supported by RRKM calculations in the zero-pressure limit suggesting the indene isomer accounted for 81–91% of all hydrogen-loss products formed5. Although the 1-phenyl-1-propyne (p2) isomer was predicted to occur at a level of 7–10%, it could not be explicitly traced in the crossed molecular beam experiments. The most recent theory describing the C6H5 plus CH3CCH reaction suggests the phenyl addition to methylacetylene is inhibited by at least a 6 kJ mol−1 entrance barrier1, and results in the formation of the geometric isomer of i2 with the methyl-group set trans to the phenyl ring that undergoes cis-trans isomerization to i2 via a barrier of only 20 kJ mol−1; all remaining transition states are energetically lower than the separated reactants and the system proceeds spontaneously to indene (p1) via annulation at the phenyl moiety. Isomerization to i1, the initial intermediate formed in the title reaction, is inhibited by a rather large barrier of 221 kJ mol−1 and thus is not competitive with the low-energy path to indene formation. Hence the nature of the entrance channel (barrier vs barrierless), initial intermediate formed, and the high-energy i1–i2 transition state (ts1) underlie the distinct reaction dynamics followed in the analogous CH3CC + C6H6 and CH3CCH + C6H5 reactions under similar experimental conditions.
Conclusion
Crossed molecular beams (CMB) experiments, combined with electronic structure and statistical calculations, were exploited to investigate the reaction dynamics of the of 1-propynyl (CH3CC; X2A1) reaction with benzene (C6H6; X1A1g) under single-collision conditions. The reaction was found to proceed indirectly and is initiated by the barrierless addition of 1-propynyl to a carbon atom on the benzene ring forming a low-lying intermediate i1 that eliminates the hydrogen atom to form 1-phenyl-1-propyne (C6H5CCCH3; p2) in an overall exoergic reaction (experimental, −157 ± 27 kJ mol−1; computational, −151 ± 6 kJ mol−1). Our RRKM analysis suggests that this pathway should account for more than 99% of C9H8 products. Interestingly, differences in the entrance channel for the title reaction as compared to phenyl (C6H5) addition to methylacetylene (CH3CCH) give rise to distinct reaction products under similar experimental conditions, with the latter forming the PAH indene (C9H8; p1) as the major hydrogen-loss product. Compared to prior experiments probing access to the C9H9 potential energy surface, such as with phenyl addition to methylacetylene and benzyl (C6H5CH2) addition to acetylene (C2H2), the reaction mechanism uncovered here is distinguished by being the first barrierless path to C9H8 where the 1-propynyl radical can disrupt aromaticity at benzene without a penalty. Despite the advantage, this pathway is not likely to compete with PAH producing reaction schemes in high temperature combustion environments where the availability of 1-propynyl radicals are likely linked to the decomposition of and/or hydrogen abstraction from the hydrocarbon methylacetylene. Under these conditions, methylacetylene is more likely to – in the case of H atom reactions – form allene via hydrogen-assisted isomerization, or react producing acetylene by methyl displacement or the propargyl radical (CH2CCH) by hydrogen abstraction, rather than form 1-propynyl by elimination or abstraction of its acetylenic hydrogen atom32,33.
The isolobal ethynyl (C2H; X2Σ+) radical, where the methyl group has been replaced by a hydrogen atom, presents an analogous chemistry to that uncovered for the 1-propynyl radical. Using the D1-ethynyl (C2D) and D6-benzene isotopologues, molecular beams experiments demonstrated the formation of D6-phenylacetylene (C6D5CCD) in a mechanism similar to the title reaction where dissociation immediately follows the barrierless formation of a low-lying C8D7 intermediate34,35. In each case, the reactivity is localized to the radical end of the alkyne, rendering the D/CH3 substituents mere spectators in the reaction coordinate. The addition/elimination reaction mechanism via a (pseudo)barrierless entrance channel is also observed in the isoelectronic boronyl (BO; X2Σ+)-benzene and cyano (CN; X2Σ+)-benzene reaction systems. Addition of the boronyl radical followed by hydrogen atom loss results in the overall exoergic formation of phenyl oxoborane (C6H5BO)36,37; also nearly 20 years before its detection in the interstellar medium38, molecular beams experiments demonstrated that cyanobenzene (C6H5CN) is formed via a low-energy, barrier-less pathway involving two neutral reactants that, in theory, proceeds spontaneously at 0 K and proposed the aromatic molecule to be present in cold molecular clouds39,40.
Although the 1-propynyl radical has not yet been detected in astrophysical sources, methylacetylene – a potential precursor – is both abundant and prolific in different regions of space41,42, and could provide a source of 1-propynyl radicals in the photodissociation region of molecular clouds or near carbon-rich AGB stars via photolytic cleavage of the acetylenic C–H bond (CH3CCH + hν → CH3CC + H)24,25,26. To establish the activity of the 1-propynyl radical in astrochemistry, laboratory characterizations of its structure are needed to aid observation efforts toward its detection and ultimately its inclusion in astrochemical modelling networks alongside its low-energy isomer, propargyl (CH2CCH). Considering that all barriers are below the energy of the separated reactants, 1-propynyl addition to benzene is a plausible reaction scheme for the low temperature extremes that persist in cold molecular clouds and strongly suggests the existence of 1-phenyl-1-propyne (C6H5CCCH3) in molecular clouds, though additional spectroscopic data are required to support its assignment among existing astronomical data43.
Methods
Experimental
The reactions of 1-propynyl (CH3CC; X2A1) with benzene (C6H6; X1A1g) and D6-benzene (C6D6; X1A1g) were carried out under single collision conditions exploiting a universal crossed molecular beams machine at the University of Hawaii44,45,46,47,48,49. The pulsed 1-propynyl radical beam was produced by photodissociation of 1-iodopropyne (CH3CCI; TCI, 99%+) seeded at a level of 0.5% in helium (99.9999%; AirGas) at 193 nm (Complex 110, Coherent, Inc.) at 30 Hz and 20 mJ per pulse in the primary source chamber50,51,52,53. This gas mixture was stored in a Teflon-coated sample cylinder and was introduced into a piezoelectric pulsed valve operating at 60 Hz, pulse widths of 80 μs, peak voltages of −400 V, and at 760 Torr backing pressure. The pulsed 1-propynyl beam passes through a skimmer and is then velocity selected by a four-slot chopper wheel rotating at 120 Hz. On-axis (Θ = 0°) characterization of the primary beam determines a peak velocity vp of 1658 ± 12 m s−1 and speed ratio S of 7.1 ± 0.3. This section of the primary beam collides perpendicularly with a supersonic beam of (D6)-benzene prepared in the secondary source chamber. A pulsed valve in the secondary source operated at a repetition rate of 60 Hz, a pulse width of 80 μs, and a peak voltage of −400 V, generated a pulsed molecular beam of (D6)-benzene [Aldrich Chemistry; ≥99.9% (>99.9% atom D)] seeded in argon (99.9999%, AirGas) at fractions of 10% at 550 Torr. The (D6)-benzene peak velocities were determined to be vp = 622 ± 10 ms−1 with S = 19.3 ± 0.6 (vp = 623 ± 9 ms−1, S = 19.3 ± 0.4) resulting in a nominal collision energy EC of 40.8 ± 0.5 kJ mol−1 (41.8 ± 0.6 kJ mol−1) and centre-of-mass angle ΘCM of 36.9 ± 0.5° (39.0 ± 0.5°) (Table 1). We note that any propargyl (CH2CCH; X2B1) radicals formed in the photodissociation of 1-iodopropyne as the result of isomerization from 1-propynyl do not react under our experimental conditions with benzene, where addition to benzene is inhibited by a barrier of 65–75 kJ mol−1 54; this barrier cannot be overcome at collision energies of 40.8 ± 0.5 kJ mol−1 in our present experiments.
The crossed molecular beams machine employs two beam sources affixed at 90 degrees and a triply differentially pumped universal detector that is rotatable in the plane defined by the reactant beams55. The detector houses an electron impact ionizer coupled to a quadrupole mass spectrometer that permits filtering of ionized (80 eV) products according to mass-to-charge ratio. Ions are ultimately registered by a photomultiplier tube and filed into bins by multichannel scaling according to time of arrival to produce a time-of-flight (TOF) spectrum. The laboratory data are forward-convoluted to the centre-of-mass (CM) frame and the resulting CM translational energy P(ET) and angular T(θ) flux distributions are analysed to inform the reaction dynamics56,57. Errors of the P(ET) and T(θ) functions are determined within the 1σ error limits of the accompanying LAB angular distribution while maintaining a good fit of the laboratory TOF spectra.
Theoretical
Geometries of the reactants, intermediates, transition states, and products of the CH3CC - C6H6 system were optimized at the density functional B3LYP/6-311 G(d,p) level of theory58,59. Vibrational frequencies were computed at the same theoretical level and were used for the evaluation of zero-point vibrational energy corrections (ZPE) and in calculations of rate constants. Energies were refined by single-point calculations using the model chemistry G3(MP2,CC)//B3LYP/6-311 G(d,p) level of theory60,61,62. This composite approach normally provides a chemical accuracy of 3–6 kJ mol–1 for the relative energies and 0.01–0.02 Å for bond lengths as well as 1–2° for bond angles62. The ab initio calculations were performed using the GAUSSIAN 0963 and MOLPRO 201064 program packages. Rate constants of all pertinent unimolecular reaction steps on the C9H9 PES following initial association of 1-propynyl with benzene were computed using Rice-Ramsperger-Kassel-Marcus (RRKM) theory65,66,67, as functions of available internal energy of each intermediate or transition state, where numbers and densities of states were obtained within the harmonic approximation using B3LYP/6–311 G(d,p) computed frequencies. The internal energy was taken as a sum of the collision energy and a negative of the relative energy of a species with respect to the reactants (the chemical activation energy). One energy level was considered throughout as at a zero-pressure limit. Then, RRKM rate constants were utilized to compute product branching ratios by solving first-order kinetic equations within steady-state approximation68 using a newly developed computer code52. Dipole and quadrupole moments and isotropic polarizabilities of CH3CC and C6H6 required for the long-range transition state theory69 calculations of the entrance channel rate constant were computed at the density functional ωB97XD level of theory70 with Dunning’s correlation-consistent cc-pVTZ basis set71.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding authors on reasonable request.
References
Zhang, F. et al. VUV photoionization study of the formation of the indene molecule and its isomers. J. Phys. Chem. Lett. 2, 1731–1735 (2011).
Kaiser, R. I., Asvany, O. & Lee, Y. T. Crossed beam investigation of elementary reactions relevant to the formation of polycyclic aromatic hydrocarbon (PAH)-like molecules in extraterrestrial environments. Planet. Space Sci. 48, 483–492 (2000).
Kaiser, R. I., Yamada, M. & Osamura, Y. A crossed beam and ab initio investigation of the reaction of hydrogen sulfide, H2S(XA1), with dicarbon molecules, C2(X1Σg +). J. Phys. Chem. A 106, 4825–4832 (2002).
Kaiser, R. I. et al. Elementary reactions of the phenyl radical, C5H5, with C3H4 isomers, and of benzene, C6H6, with atomic carbon in extraterrestrial environments. Astron. Astrophys. 406, 385–391 (2003).
Parker, D. D. S. N., Zhang, D. F., Kaiser, D. R. I., Kislov, D. V. V. & Mebel, D. A. M. Indene formation under single-collision conditions from the reaction of phenyl radicals with allene and methylacetylene—a crossed molecular beam and ab initio study. Chem. - Asian J. 6, 3035–3047 (2011).
Parker, D. S. N., Kaiser, R. I., Kostko, O. & Ahmed, M. Selective formation of indene through the reaction of benzyl radicals with acetylene. ChemPhysChem 16, 2091–2093 (2015).
Hansen, N., Schenk, M., Moshammer, K. & Kohse-Höinghaus, K. Investigating repetitive reaction pathways for the formation of polycyclic aromatic hydrocarbons in combustion processes. Combust. Flame 180, 250–261 (2017).
Kaiser, R. I. et al. Crossed beam reaction of phenyl radicals with unsaturated hydrocarbon molecules. I. Chemical dynamics of phenylmethylacetylene (C6H5CCCH3;X1A′) formation from reaction of C6H5(X 2A1) with methylacetylene, CH3CCH(X1A1). J. Chem. Phys. 112, 4994–5001 (2000).
Vereecken, L. et al. Reaction of phenyl radicals with propyne. J. Am. Chem. Soc. 124, 2781–2789 (2002).
Gu, X., Zhang, F., Guo, Y. & Kaiser, R. I. Reaction dynamics of phenyl radicals (C6H5, X2A′) with methylacetylene (CH3CCH(XA1)), allene (H2CCCH2(X1A1)), and their D4-isotopomers. J. Phys. Chem. A 111, 11450–11459 (2007).
Gu, X. & Kaiser, R. I. Reaction dynamics of phenyl radicals in extreme environments: a crossed molecular beam study. Acc. Chem. Res. 42, 290–302 (2008).
Yuan, W. et al. Experimental and kinetic modeling study of premixed n-butylbenzene flames. Proc. Combust. Inst. 36, 815–823 (2017).
Moshammer, K. et al. Aromatic ring formation in opposed-flow diffusive 1,3-butadiene flames. Proc. Combust. Inst. 36, 947–955 (2017).
Ruwe, L., Moshammer, K., Hansen, N. & Kohse-Höinghaus, K. Influences of the molecular fuel structure on combustion reactions towards soot precursors in selected alkane and alkene flames. Phys. Chem. Chem. Phys. 20, 10780–10795 (2018).
Martin, J. W. et al. Polar curved polycyclic aromatic hydrocarbons in soot formation. Proc. Combust. Inst. (2018).
Wu, X.-Z. et al. Formation of curvature subunit of carbon in combustion. J. Am. Chem. Soc. 138, 9629–9633 (2016).
Lovas, F. J. et al. Interstellar Chemistry: A strategy for detecting polycyclic aromatic hydrocarbons in space. J. Am. Chem. Soc. 127, 4345–4349 (2005).
Parker, D. S. N., Kaiser, R. I., Troy, T. P. & Ahmed, M. Hydrogen abstraction/acetylene addition revealed. Angew. Chem., Int. Ed. 53, 7740–7744 (2014).
Kaiser, R. I. Experimental investigation on the formation of carbon-bearing molecules in the interstellar medium via neutral–neutral reactions. Chem. Rev. 102, 1309–1358 (2002).
Stahl, F. et al. Reaction of the ethynyl radical, C2H, with methylacetylene, CH3CCH, under single collision conditions: implications for astrochemistry. J. Chem. Phys. 114, 3476–3487 (2001).
Kaiser, R. I. et al. Chemical dynamics of D1-methyldiacetylene (CH3CCCCD; X1A1) and D1-ethynylallene (H2CCCH(C2D); X1A′) formation from reaction of C2D(X2Σ+) with methylacetylene, CH3CCH(X1A1). J. Chem. Phys. 114, 3488–3496 (2001).
Kaiser, R. I., Stranges, D., Lee, Y. T. & Suits, A. G. Neutral-neutral reactions in the interstellar medium. I. Formation of carbon hydride radicals via reaction of carbon atoms with unsaturated hydrocarbons. Astrophys. J. 477, 982–989 (1997).
Irvine, W. M., Hoglund, B., Friberg, P., Askne, J. & Ellder, J. The increasing chemical complexity of the Taurus dark clouds: detection of CH3CCH and C4H. Astrophys. J. 248, L113–L117 (1981).
Harich, S., Lin, J. J., Lee, Y. T. & Yang, X. Photodissociation dynamics of propyne at 157 nm. J. Chem. Phys. 112, 6656–6665 (2000).
Sun, W., Yokoyama, K., Robinson, J. C., Suits, A. G. & Neumark, D. M. Discrimination of product isomers in the photodissociation of propyne and allene at 193 nm. J. Chem. Phys. 110, 4363–4368 (1999).
Ganot, Y., Rosenwaks, S. & Bar, I. H and D release in ~243.1 nm photolysis of vibrationally excited 3ν1, 4ν1, and 4νCD overtones of propyne-d3. J. Chem. Phys. 120, 8600–8607 (2004).
Jones, B. M. et al. Formation of benzene in the Interstellar Medium. Proc. Natl. Acad. Sci. USA 108, 452–457 (2011).
Levine, R. D. Molecular Reaction Dynamics. (Cambridge University Press: Cambridge, U.K., 2005).
Miller, W. B., Safron, S. A. & Herschbach, D. R. Exchange reactions of alkali atoms with alkali halides: A collision complex mechanism. Discuss. Faraday Soc. 44, 108–122 (1967).
Smith, D. J. & Grice, R. Angular distributions of reactive scattering arising from persistent complexes with asymmetric top transition states. Mol. Phys. 73, 1371–1382 (1991).
Grice, R. Dynamics of persistent collision complexes in molecular beam reactive scattering. Int. Rev. Phys. Chem. 14, 315–326 (1995).
Ryazantsev, M. N., Jamal, A., Maeda, S. & Morokuma, K. Global investigation of potential energy surfaces for the pyrolysis of C1-C3 hydrocarbons: toward the development of detailed kinetic models from first principles. Phys. Chem. Chem. Phys. 17, 27789–27805 (2015).
Rosado-Reyes, C. M., Manion, J. A. & Tsang, W. Kinetics of the thermal reaction of H atoms with propyne. J. Phys. Chem. A 114, 5710–5717 (2010).
Jones, B., Zhang, F., Maksyutenko, P., Mebel, A. M. & Kaiser, R. I. Crossed molecular beam study on the formation of phenylacetylene and its relevance to Titan’s atmosphere. J. Phys. Chem. A 114, 5256–5262 (2010).
Gu, X., Zhang, F., Guo, Y. & Kaiser, R. I. Crossed-molecular-beam study on the formation of phenylacetylene from phenyl radicals and acetylene. Angew. Chem., Int. Ed. 46, 6866–6869 (2007).
Kaiser, R. I. & Balucani, N. Exploring the gas phase synthesis of the elusive class of boronyls and the mechanism of boronyl radical reactions under single collision conditions. Acc. Chem. Res. 50, 1154–1162 (2017).
Parker, D. S. N. et al. Gas-phase synthesis of phenyl oxoborane (C6H5BO) via the reaction of boron monoxide with benzene. J. Org. Chem. 78, 11896–11900 (2013).
McGuire, B. A. et al. Detection of the aromatic molecule benzonitrile (C6H5CN) in the interstellar medium. Science 359, 202 (2018).
Balucani, N. et al. Crossed beam reaction of cyano radicals with hydrocarbon molecules. I. Chemical dynamics of cyanobenzene (C6H5CN; X1A1) and perdeutero cyanobenzene (C6D5CN; X1A1) formation from reaction of CN(X2Σ+) with benzene C6H6(X1A1g), and D6-benzene C6D6(X1A1g). J. Chem. Phys. 111, 7457–7471 (1999).
Kaiser, R. I. & Balucani, N. The formation of nitriles in hydrocarbon-rich atmospheres of planets and their satellites: laboratory investigations by the crossed molecular beam technique. Acc. Chem. Res. 34, 699–706 (2001).
Guzmán, V. V. et al. Chemical complexity in the horsehead photodissociation region. Faraday Discuss. 168, 103–127 (2014).
Gratier, P. et al. A new reference chemical composition for TMC-1. Astrophys. J., Suppl. Ser. 225, 25 (2016).
Powers, D. E., Hopkins, J. B. & Smalley, R. E. Vibrational relaxation in jet-cooled phenylalkynes. J. Chem. Phys. 74, 5971–5976 (1981).
Kaiser, R. I., Balucani, N., Charkin, D. O. & Mebel, A. M. A crossed beam and ab initio study of the C2(X1Σg +/a3Πu) + C2H2(X1Σg +) reactions. Chem. Phys. Lett. 382, 112–119 (2003).
Balucani, N., Asvany, O., Kaiser, R. I. & Osamura, Y. Formation of three C4H3N isomers from the reaction of CN (X2Σ+) with allene, H2CCCH2 (XA1), and methylacetylene, CH3CCH (X1A1): a combined crossed beam and ab initio study. J. Phys. Chem. A 106, 4301–4311 (2002).
Kaiser, R. I., Mebel, A. M., Chang, A. H. H., Lin, S. H. & Lee, Y. T. Crossed-beam reaction of carbon atoms with hydrocarbon molecules. V. Chemical dynamics of n-C4H3 formation from reaction of C(3Pj) with allene, H2CCCH2(X 1A1). J. Chem. Phys. 110, 10330–10344 (1999).
Kaiser, R. I. et al. Crossed beams reaction of atomic carbon, C(3Pj), with D6-benzene, C6D6(X 1A1g): observation of the per-deutero-1,2-didehydro-cycloheptatrienyl radical, C7D5(X2B2). J. Chem. Phys. 110, 6091–6094 (1999).
Balucani, N., Mebel, A. M., Lee, Y. T. & Kaiser, R. I. A combined crossed molecular beam and ab initio study of the reactions C2(X1Σg +, a3Πu) + C2H4 → n-C4H3(X2Aʹ) + H(2S1/2). J. Phys. Chem. A 105, 9813–9818 (2001).
Kaiser, R. I. et al. Untangling the chemical evolution of Titan’s atmosphere and surface - from homogeneous to heterogeneous chemistry. Faraday Discuss. 147, 429–478 (2010).
Thomas, A. M., Zhao, L., He, C., Mebel, A. M. & Kaiser, R. I. A combined experimental and computational study on the reaction dynamics of the 1-propynyl (CH3CC)–acetylene (HCCH) system and the formation of methyldiacetylene (CH3CCCCH). J. Phys. Chem. A 122, 6663–6672 (2018).
Thomas, A. M. et al. Combined experimental and computational study on the reaction dynamics of the 1-propynyl (CH3CC)–1,3-butadiene (CH2CHCHCH2) system and the formation of toluene under single collision conditions. J. Phys. Chem. A 123, 4104–4118 (2019).
He, C. et al. Elucidating the chemical dynamics of the elementary reactions of the 1-propynyl radical (CH3CC; X2A1) with methylacetylene (H3CCCH; X1A1) and allene (H2CCCH2; X1A1). J. Phys. Chem. A 123, 5446–5462 (2019).
He, C., Thomas, A. M., Galimova, G. R., Mebel, A. & Kaiser, R. I. Gas phase formation of methyltriacetylene (CH3(C≡C)3H) - an interstellar molecule. ChemPhysChem 20, 1912–1917 (2019).
Kislov, V. V. & Mebel, A. M. Ab initio G3-type/statistical theory study of the formation of indene in combustion flames. I. Pathways involving benzene and phenyl radical. J. Phys. Chem. A 111, 3922–3931 (2007).
Kaiser, R. I., Mebel, A. M. & Lee, Y. T. Chemical dynamics of cyclopropynylidyne (c-C3H; X2B2) formation from the reaction of C(1D) with acetylene, C2H2(X1Σg +). J. Chem. Phys. 114, 231–239 (2001).
Gu, X., Guo, Y., Zhang, F., Mebel, A. M. & Kaiser, R. I. Reaction dynamics of carbon-bearing radicals in circumstellar envelopes of carbon atars. Faraday Discuss. 133, 245–275 (2006).
Huang, L. C. L., Balucani, N., Lee, Y. T., Kaiser, R. I. & Osamura, Y. Crossed beam reaction of the cyano radical, CN(X 2Σ+), with methylacetylene, CH3CCH (X 1A1): observation of cyanopropyne, CH3CCCN (X 1A1), and cyanoallene, H2CCCHCN (X 1A′). J. Chem. Phys. 111, 2857–2860 (1999).
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988).
Curtiss, L. A., Raghavachari, K., Redfern, P. C., Rassolov, V. & Pople, J. A. Gaussian-3 (G3) theory for molecules containing first and second-row atoms, J. Chem. Phys. 109, 7764–7776 (1998).
Baboul, A. G., Curtiss, L. A., Redfern, P. C. & Raghavachari, K. Gaussian-3 theory using density functional geometries and zero-point energies, J. Chem. Phys. 110, 7650–7657 (1999).
Curtiss, L. A., Raghavachari, K., Redfern, P. C., Baboul, A. G. & Pople, J. A. Gaussian-3 theory using coupled cluster energies. Chem. Phys. Lett. 314, 101–107 (1999).
Frisch, M. et al. GAUSSIAN 09, revision A.1 (Gaussian Inc.: Wallingford, CT, 2009).
Werner, H.-J. et al. MOLPRO, Version 2010.1, A package of ab initio programs, University of Cardiff: Cardiff, UK, see http://www.molpro.net (2010).
Robinson, P. J. & Holbrook, K. A. Unimolecular Reactions. (John Wiley & Sons, Ltd.: New York, NY, 1972).
Eyring, H., Lin, S. H. & Lin, S. M. Basic Chemical Kinetics. (John Wiley and Sons, Inc.: New York, NY, 1980).
Steinfield, J., Francisco, J. & Hase, W. Chemical Kinetics and Dynamics. (Prentice Hall: Englewood Cliffs, NJ, 1982).
Kislov, V. V., Nguyen, T. L., Mebel, A. M., Lin, S. H. & Smith, S. C. Photodissociation of benzene under collision-free conditions: an ab initio/Rice-Ramsperger-Kassel-Marcus study. J. Chem. Phys. 120, 7008–7017 (2004).
Georgievskii, Y. & Klippenstein, S. J. Long-range transition state theory. J. Chem. Phys. 122, 194103 (2005).
Chai, J.-D. & Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008).
Dunning, T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989).
Acknowledgements
This work was supported by the U.S. Department of Energy, Basic Energy Sciences DE-FG02-03ER15411 and DE-FG02-04ER15570 to the University of Hawaii and to Florida International University, respectively. Ab initio calculations at Samara University were supported by the Russian Ministry of Science and Higher Education within the major project “Reaction Kinetics and Dynamics in Combustion, Astrochemistry and Astrobiology” on fundamental research in priority areas outlined by the Presidium of the Russian Academy of Sciences, whereas rate constants calculations were supported by the Grant No. 14.Y26.31.0020.
Author information
Authors and Affiliations
Contributions
A.M.T., S.D. and R.I.K. carried out the experimental measurements and data analysis. G.R.G. and A.M.M. performed the theoretical calculations. All authors discussed the results and contributed to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thomas, A.M., Doddipatla, S., Kaiser, R.I. et al. A Barrierless Pathway Accessing the C9H9 and C9H8 Potential Energy Surfaces via the Elementary Reaction of Benzene with 1-Propynyl. Sci Rep 9, 17595 (2019). https://doi.org/10.1038/s41598-019-53987-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53987-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
