
Abstract
Surface engineering has been shown critical for the success of perovskite solar cells by passivating the surface enriched defects and mobile species. The discovery of surface modulators with superior interaction strength to perovskite is of paramount importance since they can retain reliable passivation under various environments. Here, we report a chelation strategy for surface engineering of CsPbI2Br perovskite, in which dithiocarbamate molecules can be coordinate to surface Pb sites via strong bidentate chelating bonding. Such chelated CsPbI2Br perovskite can realize excellent passivation of surface under-coordinated defects, reaching a champion power conversion efficiency of 17.03% and an open-circuit voltage of 1.37 V of CsPbI2Br solar cells. More importantly, our chelation strategy enabled excellent device stability by maintaining 98% of their initial efficiency for over 1400 h in ambient condition. Our findings provide scientific insights on the surface engineering of perovskite that can facilitate the further development and application of perovskite optoelectronics.
Similar content being viewed by others
Introduction
Lead halide perovskite-based solar cells are one of the most promising photovoltaic technologies benefited from its high efficiency, low cost, and solution processibility1,2,3,4,5,6. Unfortunately, the hybrid perovskites are susceptible to be degraded under thermal condition due to the volatility nature of the organic cations7,8,9. Replacing the volatile organic cations with inorganic Cs+ can offer intrinsically thermal stable perovskite phase over 400 °C with a tunable bandgap between 1.73 eV of CsPbI3 to 2.25 eV of CsPbBr310,11. Among various cesium halide perovskites, CsPbI2Br perovskite is considered to be a good candidate for the high efficiency and stable all-inorganic perovskite solar cells (PSCs) due to its reasonable Goldschmidt tolerance factor and the lower phase transition temperature with band gaps between 1.82 and 1.92 eV12,13. Currently, the power conversion efficiency (PCE) of over 16% has been achieved for the CsPbI2Br solar cells, but their moisture and light instability is still an outstanding issue that remained to be solved14,15.
The degradation of perovskites has been identified to be initialized from the surfaces and/or grain boundaries, where are enriched with undercoordinated ions or mobile species16,17,18. These defective surfaces also introduce electronic trap states that act as fast channels for the nonradiative charge recombination19. Therefore, numerous studies have highlighted the significance of the passivation of surface defects and grain boundaries for achieving efficient and stable PSCs20,21,22,23,24,25. For example, fullerene and its derivatives can accept one electron from Lewis base type defects to passivate the perovskite surfaces in early studies26,27. Lewis bases, such as π-conjugated 6TIC-4F and theophylline molecules, have been employed to not only passivate the uncoordinated surface of the perovskite, but also better align the interfacial energy levels15,28. Most recently, oxysalts have been reported as surface inorganic passivation layers that can suppress ion migration and enhance the device stability by the merit of strong primary ionic bonding between lead cations of perovskite and sulfate anions29. In principle, the reliable and stable surface passivation for perovskites can be achieved through the formation of the strong chemical bond after passivation since its dissociation becomes difficult under various environmental stimuli. Among various bonding types, the chelation, that is embodied by a special bonding mode of polydentate molecules to central metal ions, can enable enhanced affinity to the perovskite surface than that of conventional nonchelating (monodentate) ligands30,31.
In this paper, we introduce the diethyldithiocarbamate (DDTC) molecule as a chelating agent for the surface engineering of CsPbI2Br perovskite. It was found that DDTC molecule strongly coordinates to surface Pb cation of perovskite via a bidentate chelating bonding. Such chelating structure enabled excellent and persistent passivation of surface defects of CsPbI2Br perovskite, generating significantly enhanced efficiency of 17.02% for CsPbI2Br solar cells as well as increased humidity and irradiation stability.
Results
Surface chelation of cesium halide perovskite
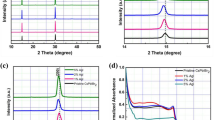
Figure 1a shows the optimized CsPbI2Br (001) surface associated to lead diethyldithiocarbamate (Pb(DDTC)2) molecule, in which two –NCS2 groups anchor at central Pb ion with a bidentate configuration (see the atomic structure in Supplementary Fig. 1)30,31. The theoretical results based on the density functional theory (DFT) reveal that the Pb(DDTC)2 is adsorbed strongly on the surface with an adsorption energy of −1.73 eV, which is much larger than that of the prevalent passivation molecules, such as ammoniums or carboxylic acids of about −0.4 and −0.3 eV32,33. As a further comparison, the adsorption energy of H2O on the CsPbI2Br (001) surface is calculated to be only about one-fifth of it (−0.32 eV), which indicates the much weaker adsorption strength of water to perovskite (Supplementary Fig. 2a). In addition, positively charged Pb atom from Pb(DDTC)2 can be bonded strongly with surface Br and I atoms to neutralize the surface charge. However, the adsorption energy of PbI2 on CsPbI2Br (001) surface was only −1.11 eV (Supplementary Fig. 2b), which suggests that the chelation between DDTC and surface undercoordinated Pb can further strengthen the adsorption. When DDTC anions bond to metal centers, they typically have serval coordination modes, such as isobidentate, anisobidentate, monodentate, triconnective (Supplementary Fig. 3)31,34. We notice that the DDTC molecules retain a bidentate chelating bonding to the center Pb cation in Pb(DDTC)2, yet offers extra interaction to another surface Pb atom, which causes the superior adsorption strength. Since the undercoordinated atoms in the pristine perovskite surface are typically the active sites for water adsorption and act as the electronic trap sites, our results suggest that the chelation of CsPbI2Br perovskite surfaces by using Pb(DDTC)2 can be a promising approach to stabilize them under humid environment and enhance the overall performance.

a Atomic structure of optimized CsPbI2Br (001) surface with Pb(DDTC)2 molecule. b ToF-SIMS depth profile of the CsPbI2Br perovskite (PVK) film with Pb(DDTC)2 (chelated CsPbI2Br), measured in negative polarity. The measured film was structured as glass/FTO/c-TiO2/perovskite. The concentration of Pb(DDTC)2 used for the chelated sample is 0.015 M. c FTIR spectra of Pb(DDTC)2 (purple) and chelated CsPbI2Br (gray) samples. The concentration of Pb(DDTC)2 and CsPbI2Br perovskite in precursor solution are 0.02 and 1.0 M, respectively. C–N stretching and C–S stretching were highlighted by blue and green colored dotted line, respectively. d Evolution of normalized absorbance for pristine (green) and chelated (purple, Pb(DDTC)2) CsPbI2Br thin films under dark in ambient atmosphere (RH, 15 ± 3%). Absorption intensities at the wavelength of 400 nm were extracted from the UV–Vis absorption spectra of CsPbI2Br films. Insets are the photographs of pristine and chelated CsPbI2Br films after exposure to ambient air for 70 and 95 days, respectively.
According to the theoretical prediction, we employed Pb(DDTC)2 as an additive in the perovskite precursor solution. The chelating agent is well dissolved in perovskite precursor solution, albeit insoluble in water (Supplementary Fig. 4). The addition of Pb(DDTC)2 with the concentration below 0.015 M almost do not change the surface morphology and roughness of the CsPbI2Br films as confirmed by the scanning electron microscopy (SEM) and the AFM images (Supplementary Figs. 5 and 6). X-ray diffraction (XRD) patterns in Supplementary Fig. 7 reveal the α-phase perovskite structure of all samples without notable shift of diffraction peaks. This implies that the added DDTC molecules might be distributed at the grain boundaries of perovskite because of its large size26. Time-of-flight secondary-ion mass spectrometry (ToF-SIMS) further shows strong H− and S− signals at the perovskite surface and the perovskite/TiO2 interface, while their intensities are less than a tenth of that at the interface throughout the perovskite film (Fig. 1b). The ToF-SIMS result presented here thus corroborates the enrichment of DDTC on the surface or interface of the polycrystalline perovskite film. In addition, the XPS results confirm the absence of Sin bare TiO2/FTO substrate (Supplementary Fig. 8). Therefore, the S− signal in the TiO2 and FTO region of ToF-SIMS spectra should be stemmed from DDTC molecules.
Fourier transform infrared (FTIR) spectra of CsPbI2Br perovskite with Pb(DDTC)2 (chelated CsPbI2Br perovskite) show similar characteristic absorption peaks with that of Pb(DDTC)2, which ascertains the existence of chelating agents in perovskite after thermal annealing at 160 °C (Supplementary Fig. 9). The FTIR absorption bands at 830–1000 cm−1, 1450–1580 cm−1, and 2850–3000 cm−1 regions primarily correspond to stretching vibration of C–S, C–N, and C–H of DDTC molecules, respectively (Fig. 1c)34,35. The C–N bond of DDTC molecules for both samples belongs to thioureide from with partial double bond character, as the v(C–N) locates between v(C=N) (1640–1690 cm−1) and v(C–N) bands (1250–1350 cm−1) (Supplementary Fig. 10)34,36. The occurrence of vas band (1067 cm−1) and vs band (980 cm−1) of –CS2 of chelated perovskites reveals the bidentate nature of the dithiocarbamate moiety with slight geometrical distortion35,36. Compared with Pb(DDTC)2, the v(C–N) and v(C–S) of the chelated CsPbI2Br perovskite shifted from 1481 and 904 cm−1 to 1497 and 910 cm−1, respectively, suggestive of the enhanced electron density near C–N bond of DDTC molecule, which is also supported by the change of Pb–S vibration peak (300–400 cm−1) from Raman spectra (Supplementary Fig. 11). Furthermore, 13C-NMR spectra in Supplementary Fig. 12 show that carbon signal of –NCS2 group of DDTC with CsPbI2Br shifted to high-field by Δδ = 2.0 ppm compared to that of pure Pb(DDTC)2, in consistent with FTIR and Raman results. This behavior might be stemmed from the electron donation from the extra unsaturated Pb sites of perovskite surface as suggested by the DFT simulation37,38,39. Orthogonal FTIR measurements further reveal the dependence of C–N stretching of DDTC molecules on the targeted materials: v(C–N) of DDTC molecules with CsI, PbI2 and CsPbI2Br are at 1481, 1495, and 1495 cm−1, respectively (Supplementary Fig. 13). The shift of v(C–N) implies that the DDTC molecules mainly coordinate to Pb ions from PbI2 or perovskite, other than Cs ions from CsI that can be explained by Pearson acid–base concept, in good consistence with 13C-NMR results.
The surface chemical states of halides of perovskite were characterized by X-ray photoelectron spectroscopy (XPS). The I 3d spectra exhibit two contributions, 3d5/2 and 3d3/2, located at 618.5 and 629.8 eV for pristine film. The Br 3d5/2 spectra exhibit two peaks at 68.4 and 69.4 eV, respectively (Supplementary Fig. 14)40. Inclusion of Pb(DDTC)2 decreases the binding energies of I 3d5/2 and Br 3d5/2 to be 618.2 and 68.2 eV, respectively, which infers enhanced electron density around surface Br/I species. This can be explained as the reduction of undercoordinated halide ions on perovskite surface through the interaction with the Pb cations in Pb(DDTC)229,40. The downshifted Pb 4f peaks, as well as the emergence of and S 2s peak of the chelated film, indicating the formation of Pb-DDTC bonding at the surface15,28. Based on above structural and electronic properties combined with the theoretical results, we conclude that the DDTC molecules chelate surface Pb cations via a bidentate configuration to passivate surface unsaturated Pb sites. Moreover, the chemical bonding between surface chelated Pb cations and surface I/Br anions should be reinforced, which may further reduce the surface undercoordinated halide related defects37.
The CsPbI2Br thin films with and without Pb(DDTC)2 were then stored in the ambient environment with relative humidity (RH) of 15 ± 3% for 95 days. The chelated CsPbI2Br film kept the original color over the entire tests, whereas the pristine one gradually turned yellow at 960 h (Supplementary Fig. 15). The UV–Vis spectra present the stable light absorbance of the chelated film in ambient air over 95 days (Fig. 1d and Supplementary Fig. 14b). Furthermore, the water contact angles of CsPbI2Br films increase from 42 to 76° after chelation, which indicates the reduction of surface hydrophilicity by surface chelation (Supplementary Fig. 16).
Photovoltaic performance
The cross-sectional SEM image of the typical planar heterojunction solar cell device architecture of glass/fluorine-doped tin oxide (FTO)/compact TiO2 (c-TiO2)/CsPbI2Br/P3HT/Ag is shown in Fig. 2a. The current density–voltage (J–V) curves of CsPbI2Br devices with different concentration of Pb(DDTC)2 measured under simulated AM 1.5G illumination are shown in the Fig. 2b. The corresponding photovoltaic parameters are summarized in Supplementary Table 1. The pristine device delivered a short-circuit current density (JSC) of 15.88 mA cm−2, an open-circuit voltage (VOC) of 1.19 V, a fill factor (FF) of 77.28% and a PCE of 14.54%. The addition of Pb(DDTC)2 mainly enhanced the VOC of devices. The VOC of the CsPbI2Br devices with 0.005, 0.010, 0.015, and 0.020 M Pb(DDTC)2 molecules were 1.28, 1.31, 1.34, and 1.30 V, generating high PCEs of 15.82%, 16.34%, 16.57%, and 15.31%, respectively. The reduction of PCE with excess Pb(DDTC)2 molecules can be explained by the large series resistance of insulating molecules. Given that VOC are determined by quasi-Fermi level splitting under light irradiation, the chelated device should adopt much higher carrier concentration at open circuit probably because of the defect passivation effect of DDTC molecules. By using 0.015 M Pb(DDTC)2, we improved the average PCE from 13.70 ± 0.45% for pristine devices to 16.10 ± 0.44% for chelated ones (Fig. 2c). The champion chelated device showed a VOC of 1.34 V, a JSC of 15.78 mA cm−2, and a FF of 80.52%, generating a high PCE of 17.03% without notable hysteresis (Fig. 2d and Supplementary Table 2). This champion device is among the best reported CsPbI2Br ones (Supplementary Table 3). Moreover, a record VOC of 1.37 V for chelated CsPbI2Br cell was also attained with the VOC deficit of about 0.51 eV (Supplementary Fig. 17). The stabilized power output of champion device was recorded as 17.01% at maximum power point (MPP) under a bias of 1.11 V, in conjunction with the stable JSC of 15.32 mA cm−2 after 1000 s (Fig. 2e). The integrated current density JSC value obtained from the external quantum efficiency (EQE) measurements carried out on this device was calculated to be 15.43 mA cm−2, well matches to that obtained from J–V curve (Fig. 2f).

a Cross-sectional SEM image of a chelated perovskite device. Scale bar is 300 nm. b J–V curves of perovskite solar cells with different concentrations of Pb(DDTC)2 under simulated AM 1.5G illumination (100 mW cm−2). c PCE histograms of pristine (blue) and chelated (orange) CsPbI2Br PSCs. d J–V characteristics of the champion device for chelated CsPbI2Br solar cell at opposite scan directions. e Steady-state power output (blue) and current density (orange) of the champion cell measured at a fixed maximum power point (MPP) voltage (1.11 V) as a function of time. f EQE spectra (orange) and corresponding integrated photocurrent (blue) of the champion device.
Steady-state photoluminescence (PL) and time-resolved PL (TRPL) measurements were operated, which are typical methods to assess the electronic passivation effect of chelating molecules on CsPbI2Br films4,29,41. As shown in Fig. 3a, the PL intensity of chelated film is enhanced to ∼2.1 times larger than that of pristine one, indicating the suppression of nonradiative charge recombination25. Meanwhile, the slight blue-shift of PL peak for the chelated film confirms the reduced shallow defects. TRPL spectra of both samples manifested a biexponential decay composed of a fast and a slow component that are typically assigned to charge trapping process and carrier recombination process, respectively (Fig. 3b)23. The fast decay lifetime (τ1) and slow decay lifetime (τ2) are 11.1 and 68.2 ns for the pristine CsPbI2Br film, and 13.3 and 91.3 ns for the chelated one, respectively. The high PL intensity and prolonged lifetime of the chelated film are associated with the decrease of electronic trap density42. The lower dark current density of chelated device further indicates the reduction of shunt pathways, which may originate from the defective grain boundaries (Supplementary Fig. 18). To further evaluate the recombination behavior of the solar cell devices, the dependence of VOC on light intensities was measured and the results were plotted as a function of light intensity in logarithm scales as shown in Fig. 3c. Ideality factor (n) of the devices can be deduced by the slope of VOC as a function of light intensity (I) according to the following equation15:
where Eg is the bandgap of light absorber, k is the Boltzmann constant, T is the absolute temperature, q is the electron charge, and I0 is a constant with the same unit as I. Figure 3c shows the slopes of 1.76 kT/q for the chelated device and 2.22 kT/q for the pristine device, indicating the suppressed trap-induced recombination of the chelated device. Thermal admittance spectroscopy, as a well-established measurement of device trap density, was operated to measure the trap density of states (tDOS) of the as-fabricated devices29. As evidenced by Fig. 3d, the tDOS of chelated device are more than ten times lower than the pristine one in the deep trap region (greater than 0.50 eV). We speculate that the DDTC molecule passivate the surface undercoordinated Pb and halide ions as discussed above. Electrochemical impedance spectroscopy (EIS) characterization was performed to further probe the carrier dynamics of CsPbI2Br devices. The equivalent circuit model and fitting parameters of the impedance spectroscopy are showed in the Supplementary Fig. 1943,44. The recombination resistance (Rrec) for the chelated device is about 4.3 times of that for pristine device at bias voltage of 1.0 V (Fig. 3e). The higher Rrec of the chelated device under different bias voltages further represents the longer carrier lifetime of the chelated device, benefiting from the reduction of surface trap states (Fig. 3f)21,44.

a, b Steady-state PL and TRPL spectra, respectively, of the pristine (blue) and chelated (orange) CsPbI2Br film deposited on glass substrate. c Plots of light intensity dependent VOC of the pristine (blue) and chelated (orange) CsPbI2Br devices. d tDOS of the pristine (blue) and chelated (orange) CsPbI2Br devices extracted from thermal admittance spectroscopy. e Nyquist plots of the pristine (blue) and chelated (orange) CsPbI2Br devices under dark with a bias voltage of 1.0 V. f Dependence of Rrec on applied bias voltage for pristine (blue) and chelated (orange) CsPbI2Br device. Rrec was obtained by fitting the EIS spectra at different voltages.
Long-term device stability
Furthermore, the long-term stabilities of solar cell devices were tested under different conditions by adopting the device configuration of FTO/c-TiO2/perovskite/P3HT/Au. As given in Fig. 4a, the humid stability of chelated device was noticeably enhanced, maintaining over 98% of its original PCE under ambient conditions with 15 ± 3% humidity for 1440 h. In striking contrast, the pristine device only retained 53% of its initial efficiency within 840 h under the same conditions. The operational stability of CsPbI2Br devices presented in Fig. 4b indicated that there was only 11% efficiency drop for the chelated device after 520 h of continuous one-sun illumination in nitrogen atmosphere. As comparison, the pristine device lost 45% of its initial efficiency within 370 h. Moreover, the chelated device showed a slow decrease of PCE over time and maintained 90% of its initial efficiency after 450 h at 85 °C, while the PCE value of the pristine CsPbI2Br device dropped dramatically to 57% for 360 h (Supplementary Fig. 20). Above results have verified the essential role of Pb(DDTC)2 that stabilize the perovskite device from the degradation. Ions migration has been proved to be as a possible origin of devices’ intrinsic degradation under the working condition, which can be detected by PL emission under continuous light irradiation15,26. The PL emission peak of pristine CsPbI2Br thin film had a red shift of about 3 nm under the continuous light illumination for 10 min (Fig. 4c). Encouragingly, we found that the chelated film displayed no change in PL emission, suggesting the suppressed ion migration after chelation (Fig. 4d).

a Evolution of power conversion efficiency of the pristine (blue) and chelated (orange) CsPbI2Br devices without encapsulation. The devices were stored under ambient atmosphere with controlled humidity (RH, 15 ± 3%). b Long-term stability of the pristine (blue) and chelated (orange) devices under continuous 100 mW cm−2 white LED illumination in nitrogen glovebox. All devices configurations are FTO/c-TiO2/perovskite/P3HT/Au. Normalized PL spectra of the (c) pristine and (d) chelated CsPbI2Br films under continuous one sun equivalent illumination for 10 min. All perovskite films were deposited on glass substrates.
Discussion
In summary, we have demonstrated the effectiveness of chelating molecular modulation on the surface property of perovskite films. Our experimental and theoretical results have elucidated the detailed surface chelation interactions of DDTC molecules to the surface Pb cations, which can passivate both undercoordinated surface Pb and I/Br defects. The champion PCE and VOC of chelated CsPbI2Br cells attained 17.03% and 1.37 V, respectively, which are among the best reported results. Considering the strong adsorption of chelating molecules on perovskite, the stable bonding of chelating agents to the surface enables the long-term stability of perovskite devices. Other chelating agents, such as oxalic acid and ethylenediaminetetraacetic acid, were also tested with positive results. Our findings offer scientific avenues toward efficient and stable perovskite-based photovoltaic as well as other optoelectronic applications.
Methods
Materials and solution preparation
Lead nitrate (Pb(NO3)2, 99%), ethanol (≥99.7%) and sodium N,N-diethyldithiocarbamate trihydrate (NaDDTC·3H2O, 99%) were purchased from Sinopharm Chemical Reagent Company. Lead bromide (PbBr2, 99.9%), lead iodide (PbI2, 99.9%), cesium iodide (CsI, 99.9%), dimethyl sulfoxide (DMSO, anhydrous, ≥99.8%), dimethyl sulfoxide-d6 (DMSO-d6, ≥99.9%), chlorobenzene (≥99.9%), acetonitrile (≥99.8%), 4-tert-butylpyridine (t-BP, ≥96.0%), bis(trifluoromethane)sulfonimide lithium salt (Li-TFSI, ≥98.0%) and titanium(IV) chloride (TiCl4, >98%) were purchased from Sigma–Aldrich. Poly (3-methylthiophene) (P3HT) was purchased from Xi’an Polymer Light Technology Corp. All the chemicals and solvents were used as received without further purification. The Pb(DDTC)2 was synthesized by adding 20.0 M NaDDTC·3H2O aqueous solution of Pb(NO3)2 aqueous solution (10.0 M) dropwise under continuous magnetic stirring at room temperature. After stirring for 30 min, the yellow precipitate was filtered and washed four times with pure water and dried in an oven at 60 °C. Firstly, 30.22 mg Pb(DDTC)2 was dissolved into 2.0 mL DMSO solution to form Pb(DDTC)2-DMSO solution (0.03 M). Then, 235.5 mg PbI2, 187 mg PbBr2, and 272 mg CsI were dissolved in DMSO and Pb(DDTC)2-DMSO mixed solution to a final volume of 1 mL with desired Pb(DDTC)2 concentration, followed by stirring at room temperature overnight without filtration in a N2 glovebox.
Device fabrication and characterization
FTO substrate (8 Ω per square, Nippon Sheet Glass) was sequentially cleaned with detergent, deionized water, acetone, and alcohol under sonication. Dry substrate was then immersed in a 25 mM TiCl4 aqueous solution for 60 min at 70 °C and washed with deionized water and ethanol, followed by annealing at 500 °C for 60 min in muffle to form a c-TiO2 blocking layer. The CsPbI2Br layer was fabricated by spin-coating the precursor solution on the FTO/c-TiO2 substrate via a two-step process with 500 rpm for 3 s, and 3500 rpm for 30 s. Subsequently, these films were annealed at 43 °C for 2 min, and then at 160 °C for 10 min45. P3HT (15 mg/mL in chlorobenzene, with 45 µL Li-TFSI solution (2 mg in 1 mL acetonitrile) and 10.2 µL t-BP) was spin-coated onto the CsPbI2Br perovskite film at 2500 rpm for 25 s as a hole transporting layer. Finally, the device was finished by evaporating Ag or Au layers29.
FTIR spectroscopy, Raman spectroscopy (Raman) and Carbon-13 nuclear magnetic resonance (13C-NMR) were performed by FTIR Nicolet 6700, Laser Raman InVia Reflex and Bruker Avance 500, respectively. The surface morphology and roughness of films were characterized by field emission scanning electron microscopy (HITACHI S4800) and atomic force microscopy (AFM, Veeco/DI). The XPS measurements were performed in ESCALAB 250Xi (Mg anode, 250 W, 14 kV), and the binding energy of the C 1s peak at 284.8 eV was taken as an internal pristine. XRD spectra of the prepared perovskite films were measured using powder X-ray diffraction (PXRD, Bruker Advance D8 X-ray diffractometer, Cu Kα radiation, 40 kV). Time-of-flight secondary-ion mass spectrometry (ToF-SIMS VI, IONTOF GmbH, Muenster, Germany) elemental depth profiling was used to probing the distribution of elements. UV–Vis spectra were collected using a Cary 500 UV–Vis–NIR spectrophotometer in air ambient environments. The steady-state PL and TRPL spectra of perovskite films were acquired by Fluorolog-3-p spectrophotometer and Endinburgh FLS890 spectrometer in air at room temperature, respectively. PL excitation wavelength was 380 nm. Solar cells were illuminated by a solar light simulator (Solar IV-150A, Zolix) and light intensity was calibrated by a standard Newport calibrated KG5-filtered Si reference cell. The J–V curves of devices were measured with Keithley 2400 digital sourcemeter under AM 1.5G irradiation (100 mW cm−2) at a scan rate of 0.15 V s−1 (voltages scan range: 0.3–1.5 V, voltage step of 10 mV) in air ambient environments. Devices were masked with a metal aperture to define the active area to be 0.0625 cm2. The steady-state photocurrent output of the best-performing devices was measured by biasing the device at MPP in air ambient environments. EIS were measured out using an electrochemical workstation (Parstat 2273, Princeton) in the frequency range of 1 MHz and 1 Hz under different positive bias voltages at dark conditions in air. The EQE was carried out on a Newport-74125 system (Newport Instruments) in air.
The CsPbI2Br films and unsealed devices were stored in air ambient environments with 15 ± 3% relatively humidity for long-term humidity test. White LED light illumination of 100 mW cm−2 for illumination stability tests of unencapsulated devices in nitrogen glovebox. The unencapsulated devices were baked on the hot plate at 85 °C in nitrogen glovebox for thermal stability test.
Computational details
The first-principles DFT calculations were performed using the Vienna Ab initio Simulation Package code in this study46,47,48. The projector augmented wave method was used to describe the electron-ion interaction49, with valence electron structures of 5s25p66s1 for Cs (Cs_sv), 5d106s26p2 for Pb (Pb_d), 5s25p5 for I, 4s24p5 for Br, 3s23p4 for S, 2s22p2 for C, 2s22p3 for N, and 1s1 for H. The exchange and correlation effects of the electron–electron were described by using the Perdew–Burke–Ernzerhof (PBE) functional of a generalized gradient approximation method50. The plane-wave kinetic energy cutoff was set at 520 eV to expand the smooth part of wave functions. All calculations were carried out on the thermodynamically stable (001) surface using a (1 × 2) five-layer slab model with the periodic boundary condition. A vacuum region of 20 Å was used to avoid the interaction between slabs. The Brillouin zone was sampled using Gamma-centered (5 × 3 × 1) k-point mesh for all surface calculations. Since traditional DFT calculations at the PBE level cannot correctly include the nonlocal van der Waals interactions, the calculations with dispersion corrections may affect the adsorption energies of small molecules51,52. In this regard, the DFT‐D3 method was adopted for dispersion corrections here53. During the geometry optimizations, the bottom two layers were fixed at the bulk position, whereas the top three atomic layers and the adsorbents were fully relaxed with the energy and force convergences <10−5 eV and 0.01 eV/Å, respectively.
The adsorption energy of each adsorbate [ΔEad (eV)] was calculated as follows:
where Ead, Esurf, and Ead/surf are the energies of an adsorbate, the clean (001) surface and the surface with adsorbates, respectively. For the calculations of adsorbate molecules, a (20 × 20 × 20) Å3 unit cell and a Γ-only k-point grid were used.
Data availability
The data that support the plots within this paper are available in separate Supplementary Source Data Files in Supplementary information section. All other relevant data are available from the corresponding authors upon reasonable request.
References
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
Kim, H.-S. et al. Lead iodide perovskite sensitized all-solid-state submicron thin film mesoscopic solar cell with efficiency exceeding 9%. Sci. Rep. 2, 591 (2012).
Wehrenfennig, C., Eperon, G. E., Johnston, M. B., Snaith, H. J. & Herz, L. M. High charge carrier mobilities and lifetimes in organolead trihalide perovskites. Adv. Mater. 26, 1584–1589 (2013).
Turren-Cruz, S.-H., Hagfeldt, A. & Salibal, M. Methylammonium-free, high-performance, and stable perovskite solar cells on a planar architecture. Science 362, 449–453 (2018).
Song, J. et al. Monolayer and few‐layer all‐inorganic perovskites as a new family of two‐dimensional semiconductors for printable optoelectronic devices. Adv. Mater. 28, 4861–4869 (2016).
Chen, Z. et al. Single-crystal MAPbI3 perovskite solar cells exceeding 21% power conversion efficiency. ACS Energy Lett. 4, 1258–1259 (2019).
Bryant, D. et al. Light and oxygen induced degradation limits the operational stability of methylammonium lead triiodide perovskite solar cells. Energy Environ. Sci. 9, 1655–1660 (2016).
Domanski, K. et al. Migration of cations induces reversible performance losses over day/night cycling in perovskite solar cells. Energy Environ. Sci. 10, 604–613 (2017).
Yang, S. et al. Functionalization of perovskite thin films with moisture-tolerant molecules. Nat. Energy 1, 15016 (2016).
Duan, J. et al. Carbon-electrode-tailored all-inorganic perovskite solar cells to harvest solar and water-vapor energy. Angew. Chem. Int. Ed. 57, 5746–5749 (2018).
Tian, J. et al. Dual interfacial design for efficient CsPbI2Br perovskite solar cells with improved photostability. Adv. Mater. 31, 1901152 (2019).
Zeng, Q. et al. Inorganic CsPbI2Br perovskite solar cells: the progress and perspective. Sol. RRL 3, 1800239 (2019).
Chen, W. et al. Precise control of crystal growth for highly efficient CsPbI2Br perovskite solar cells. Joule 3, 191–204 (2019).
Xiang, W. et al. Ba-induced phase segregation and band gap reduction in mixed-halide inorganic perovskite solar cells. Nat. Commun. 10, 4686 (2019).
Wang, J. et al. Highly efficient all-inorganic perovskite solar cells with suppressed non-radiative recombination by a Lewis base. Nat. Commun. 11, 177 (2020).
Yuan, Y. et al. Anomalous photovoltaic effect in organic-inorganic hybrid perovskite solar cells. Sci. Adv. 3, e16021642 (2017).
Fan, Z. et al. Layer-by-layer degradation of methylammonium lead tri-iodide perovskite microplates. Joule 1, 548–562 (2017).
Ball, J. M. & Petrozza, A. Defects in perovskite-halides and their effects in solar cells. Nat. Energy 1, 16149 (2016).
Sherkar, T. S. et al. Recombination in perovskite solar cells: significance of grain boundaries, interface traps, and defect ions. ACS Energy Lett. 2, 1214–1222 (2017).
Kim, B., Kim, M., Lee, J. H. & Seok, S. II Enhanced moisture stability by butyldimethylsulfonium cation in perovskite solar cells. Adv. Sci. 7, 1901840 (2019).
Wu, X. et al. Stable triple cation perovskite precursor for highly efficient perovskite solar cells enabled by interaction with 18C6 stabilizer. Adv. Funct. Mater. 30, 1908613 (2019).
Wang, Y. et al. Thermodynamically stabilized β-CsPbI3-based perovskite solar cells with efficiencies >18%. Science 365, 591–595 (2019).
Chen, P. et al. In situ growth of 2D perovskite capping layer for stable and efficient perovskite solar cells. Adv. Funct. Mater. 28, 1706923 (2018).
Zeng, Q. et al. Polymer‐passivated inorganic cesium lead mixed‐halide perovskites for stable and efficient solar cells with high open‐circuit voltage over 1.3 V. Adv. Mater. 30, 1705393 (2018).
Zhang, Y. et al. High efficiency (16.37%) of cesium bromide—passivated all-inorganic CsPbI2Br perovskite solar cells. Sol. RRL 3, 1900254 (2019).
Shao, Y., Xiao, Z., Bi, C., Yuan, Y. & Huang, J. Origin and elimination of photocurrent hysteresis by fullerene passivation in CH3NH3PbI3 planar heterojunction solar cells. Nat. Commun. 5, 5784 (2014).
Zhang, M. et al. Reconfiguration of interfacial energy band structure for high-performance inverted structure perovskite. Nat. Commun. 10, 4593 (2019).
Wang, R. et al. Constructive molecular configurations for surface-defect passivation of perovskite photovoltaics. Science 366, 1509–1513 (2019).
Yang, S. et al. Stabilizing halide perovskite surfaces for solar cell operation with wide-bandgap lead oxysalts. Science 365, 473–478 (2019).
Caruso, F., Chan, M.-L. & Rossi, M. A short Pb…Pb separation in the polymeric compound bis-(pyrrolidinedithiocarbamato)lead(II) and a conformational pathway interconversion for the “Pb(II)S4” framework. Inorg. Chem. 36, 3609–3615 (1997).
Hogarth, G. Metal-dithiocarbamate complexes: chemistry and biological activity. Mini-Rev. Med. Chem. 12, 1202–1215 (2012).
Kim, B., Kim, M., Lee, J. H. & Seok, S. II Enhanced moisture stability by butyldimethylsulfonium cation in perovskite solar cells. Adv. Sci. 7, 1901840 (2019).
Zhang, L., Yu, F., Chen, L. & Li, J. Adsorption of molecular additive onto lead halide perovskite surfaces: a computational study on Lewis base thiophene additive passivation. Appl. Surf. Sci. 443, 176–183 (2018).
Tiwari, S., Reddy, K. V., Bajpai, A., Khare, K. & Nagaraju, V. Synthesis and characterization of bisdithiocarbamates from weak nitrogen bases and its metal complexes. Int. Res. J. Pure Appl. Chem. 7, 78–91 (2015).
Onwudiwe, D. C. & Ajibade, P. A. Synthesis, characterization and thermal studies of Zn(II), Cd(II) and Hg(II) complexes of N-Methyl-N-Phenyldithiocarbamate: the single crystal structure of [(C6H5)(CH3)NCS2]4Hg2. Int. J. Mol. Sci. 12, 1964–1978 (2011).
Sathiyaraj, E. & Thirumaran, S. Synthesis and spectral studies on Pb(II) dithiocarbamate complexes containing benzyl and furfuryl groups and their use as precursors for PbS nanoparticles. Spectrochim. Acta Part A: Mole. Biomol. Spectrosc. 97, 575–581 (2012).
Zuo, L. et al. Polymer-modified halide perovskite films for efficient and stable planar heterojunction solar cells. Sci. Adv. 3, e1700106 (2017).
Chen, B. et al. Imperfections and their passivation in halide perovskite solar cells. Chem. Soc. Rev. 48, 3842–3867 (2019).
Abate, A. et al. Supramolecular halogen bond passivation of organic-inorganic halide perovskite solar cells. Nano Lett. 14, 3247–3254 (2014).
Subhani, W.S., Wang, K., Du, M., Wang, X. & Liu, S. (Frank). Interface-modification-induced gradient energy band for highly efficient CsPbIBr2 perovskite solar cells. Adv. Energy Mater. 9, 1803785 (2019).
Stranks, S. D. et al. Recombination kinetics in organic-inorganic perovskites: excitons, free Charge, and subgap states. Phys. Rev. Appl. 2, 034007 (2014).
Zhu, H. et al. Tailored amphiphilic molecular mitigators for stable perovskite solar cells with 23.5% efficiency. Adv. Mater. 32, 1907757 (2020).
Ye, S. et al. A breakthrough efficiency of 19.9% obtained in inverted perovskite solar cells by using an efficient trap state passivator Cu(thiourea)I. J. Am. Chem. Soc. 139, 7504–7512 (2017).
Kim, H.-S. et al. High efficiency solid-state sensitized solar cell-based on submicrometer rutile TiO2 nanorod and CH3NH3PbI3 perovskite sensitizer. Nano Lett. 13, 2412–2417 (2013).
Liu, C. et al. All-inorganic CsPbI2Br perovskite solar cells with high efficiency exceeding 13%. J. Am. Chem. Soc. 14, 3825–3828 (2018).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 48, 13115–13118 (1993).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Klimeš, J. & Michaelides, A. Perspective: advances and challenges in treating van der waals dispersion forces in density functional theory. J. Chem. Phys. 137, 120901 (2012).
Sun, T., Wang, Y., Zhang, H., Liu, P. & Zhao, H. Adsorption and oxidation of oxalic acid on anatase TiO2 (001) surface: a density functional theory study. J. Colloid Interface Sci. 454, 180–186 (2015).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Acknowledgements
This work was financially supported by National Natural Science Funds for Distinguished Young Scholar (51725201), National Natural Science Foundation of China (51972111, 51602103, 51902185), Young Elite Scientists Sponsorship Program by CAST (2017QNRC001), International (Regional) Cooperation and Exchange Projects of the National Natural Science Foundation of China (51920105003), Innovation Program of Shanghai Municipal Education Commission (E00014), the Fundamental Research Funds for the Central Universities (JKD012016025, JKD012016022), and Shanghai Engineering Research Center of Hierarchical Nanomaterials (18DZ2252400). This research was also undertaken on the supercomputers at the National Computational Infrastructure (NCI) in Canberra, Australia, which is supported by the Australian Commonwealth Government, and Pawsey Supercomputing Centre in Perth with funding from the Australian Government and the Government of Western Australia.
Author information
Authors and Affiliations
Contributions
S.Y. and H.G.Y. directed the scientific research for this work. J.H. and S.Y. devised the idea of the project. J.H. performed the material preparation, device fabrication, and characterizations. Y.W. and J.L. conducted DFT calculations and wrote part of the paper (calculations). J.H. and S.Y. analyzed the data and prepared the paper with the help of Y.W. and H.Y. All the authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Giulia Grancini and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, J., Liu, J., Hou, Y. et al. Surface chelation of cesium halide perovskite by dithiocarbamate for efficient and stable solar cells. Nat Commun 11, 4237 (2020). https://doi.org/10.1038/s41467-020-18015-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-18015-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
