
Abstract
Senescent cells may promote tumour progression through the activation of a senescence-associated secretory phenotype (SASP), whether these cells are capable of initiating tumourigenesis in vivo is not known. Expression of oncogenic β-catenin in Sox2+ young adult pituitary stem cells leads to formation of clusters of stem cells and induction of tumours resembling human adamantinomatous craniopharyngioma (ACP), derived from Sox2− cells in a paracrine manner. Here, we uncover the mechanisms underlying this paracrine tumourigenesis. We show that expression of oncogenic β-catenin in Hesx1+ embryonic precursors also results in stem cell clusters and paracrine tumours. We reveal that human and mouse clusters are analogous and share a common signature of senescence and SASP. Finally, we show that mice with reduced senescence and SASP responses exhibit decreased tumour-inducing potential. Together, we provide evidence that senescence and a stem cell-associated SASP drive cell transformation and tumour initiation in vivo in an age-dependent fashion.
Similar content being viewed by others


Introduction
Cellular senescence defines a state of stable and long-term loss of proliferative capacity, but with retention of normal metabolic activity and viability1. The activation of the senescence programme acts as a potent tumour suppression mechanism through the activation of the p53 pathway and expression of cell cycle inhibitors (e.g. p21 (CDKN1A) and p16 (CDKN2A))2, 3. The mitogenic stimuli caused by the expression of several oncogenic proteins, including mutant β-catenin, BRAFV600E or KRASG12D, trigger DNA replication stress leading to DNA damage, activation of a DNA damage response (DDR) and the induction of senescence (named oncogene-induced senescence, OIS)4, 5. As a result, senescent cells activate a molecular programme characterised by the expression and secretion of a multitude of growth factors, matrix proteases and pro-inflammatory proteins collectively referred to as the senescence-associated secretory phenotype (SASP)6. The composition and intensity of the SASP response can be affected by factors such as the senescence-inducing mechanism, cell type and time passed since senescence initiation, suggesting that the SASP is not a singular state7,8,9,10. The activation of the SASP requires a persistent DDR and is mediated by the NF-κB and C/EBPβ pathways11. SASP-associated cytokines, IL-6 and IL-8, reinforce the senescence growth arrest, at least in some senescent cells12, 13, which is beneficial in cancer suppression. However, the paracrine activities of senescent cells through SASP activation can also promote tumourigenesis. Prominent or persistent SASP activation has been shown to: (1) disrupt cell–cell adhesion and induce epithelial-to-mesenchymal transition and invasiveness14, 15; (2) cause local inflammation12, 16; (3) modify tissue architecture17, 18; (4) facilitate development of hepatic cancer after carcinogen exposure19, 20; (5) stimulate proliferation of nearby pre- and malignant cells both in vitro21 and in vivo when co-injected with senescent cells in xenograft mouse models17, 18, 22 and (6) favour the emergence of tumour-initiating cells in cell culture models23,24,25,26. This bulk of evidence demonstrates a pro-tumourigenic role for the SASP, but whether the SASP can induce cell transformation and tumour initiation of non-tumorigenic cells in vivo remain less clear.
We have previously shown that the expression of a degradation-resistant form of β-catenin in Rathke’s pouch, the embryonic primordium of the anterior pituitary gland (Hesx1Cre/+;Ctnnb1lox(ex3)/+ mice)27, or in Sox2+ adult pituitary stem cells (Sox2CreERT2/+; Ctnnb1lox(ex3)/+ mice)28 leads to the formation of tumours that resemble human adamantinomatous craniopharyngioma (ACP). Interestingly, targeting expression of this mutant β-catenin to cell-lineage progenitors or differentiated cells in the developing pituitary is not tumourigenic, suggesting that the oncogenic effect requires an undifferentiated stem/cell precursor27. ACPs are clinically aggressive tumours associated with high morbidity and significant premature mortality29. Most human ACPs carry mutations in β-catenin leading to the over-activation of the WNT/β-catenin pathway30,31,32,33. In agreement with this finding, cells showing nucleo-cytoplasmic accumulation of β-catenin and activation of the WNT pathway are present in mouse and human tumours, commonly grouped in whorl-like structures, named ‘cell clusters’, near the invasive front29. These cell clusters are not found in any other type of pituitary tumours34, express stem cell markers27, 35 and have been proposed to play a critical role in controlling the infiltrative behaviour of surrounding tumour cells36.
Although murine clusters derive from mutant Sox2+; S100B+ adult pituitary stem cells expressing oncogenic β-catenin28, this population is not the cell-of-origin of the tumours, suggesting a non-cell autonomous role during tumourigenesis. Currently, the molecular and cellular mechanisms underlying the pro-tumorigenic role of this peculiar cell population remain to be discovered. In this study, we demonstrate through molecular and genetics approaches that murine and human clusters are functionally equivalent structures, which show a molecular signature of cellular senescence and a SASP. Our results indicate that tumour induction only occurs in the presence of robust SASP activation, therefore providing evidence for a role of senescence and SASP in tumour initiation in vivo.
Results
Pituitary embryonic precursors can induce paracrine tumours
We previously showed that the expression of oncogenic β-catenin in Sox2+ pituitary stem cells in Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ mice at 4–6 weeks of age results in the formation of pituitary tumours in a non-cell autonomous manner28. We sought to investigate whether a similar mechanism may underlie the development of pituitary tumours in Hesx1Cre/+;Ctnnb1lox(ex3)/+;R26YFP/+ mice, another model for human ACP, in which oncogenic β-catenin is expressed in the embryonic pituitary precursors that normally give rise to all the cell lineages of the mature pituitary gland, including Sox2+ stem cells27.
The pituitary gland of the Hesx1Cre/+;Ctnnb1lox(ex3)/+;R26YFP/+ mice at 18.5 dpc was enlarged in comparison with the Hesx1Cre/+;R26YFP/+ controls, but did not develop tumours at this stage and will be hereafter referred to as a ‘pre-tumoural’ pituitary (Fig. 1a). The vast majority of the cells in the developing anterior pituitary were YFP+ with abundant Ki67 expression, as expected at this developmental stage (Fig. 1c). After a variable latency period of several weeks (median age of 17.7 weeks, range 8.3–35.3 weeks37), the YFP+ population was progressively replaced by actively proliferating YFP−;Ki67+ cells (Fig. 1b, c). Eventually, in fully developed tumours (i.e. when mice reached the humane end-point), none or only scarce YFP+ cells could be observed in the periphery of the majority of the tumours (8 out of 11), which were comprised mostly of YFP− cells. Notably, Ki67 expression was mostly observed in YFP− cells at this point (Fig. 1c). Similar results were obtained from Hesx1Cre/+;Ctnnb1lox(ex3)/+;R26mTmG/+ mice, in which 4 out of 5 tumours contained large tdTomato+ areas, indicating that tumour cells were not recombined (Supplementary Fig. 1). β-catenin-accumulating cell clusters were evident at pre-tumoural stages and as expected were YFP+ (Fig. 1c) and Ki67−27. As tumours developed, β-catenin expression was elevated in YFP− cells and clusters were no longer identifiable (e.g. after 20 weeks) (Fig. 1c). Additionally, YFP− cells were synaptophysin-, suggesting that these are not neuroendocrine cells (Fig. 1c).

Tumours in the Hesx1Cre/+;Ctnnb1lox(ex3)/+;R26YFP/+ mice form non-cell autonomously after a latency period. a Brightfield and epifluorescence images of 18.5 dpc pituitaries. Note that most of the anterior lobe (al), but not the posterior (pl), is fluorescent in both the Hesx1Cre/+;R26YFP/+ control and Hesx1Cre/+;Ctnnb1lox(ex3)/+;R26YFP/+ pituitaries. The mutant pre-tumoural pituitary is enlarged relative to controls. Scale bar: 1 mm. b Brightfield and epifluorescence pictures of a pre-tumoural pituitary and tumours in Hesx1Cre/+;Ctnnb1lox(ex3)/+;R26YFP/+ mutant mice at 5, 20 and 35 weeks of age. Most of the tumour cells are not fluorescent and the YFP+ population (white arrow) is displaced to the periphery of the growing tumour mass (white asterisks). Scale bar: 5 mm. c Histological characterisation of pre-tumoural pituitaries and developing tumours from 18.5 dpc to 35 weeks postnatally. At 18.5 dpc, most of the embryonic pituitary is comprised of YFP+ cells (revealed with anti-GFP immunostaining), but there are islands of YFP− cells (DAPI+ areas). Abundant Ki67+ cells are distributed throughout the pituitary at this stage. At 5 weeks of age, the pre-tumoural pituitary remains mostly quiescent with very few Ki67+ cells. Note the presence of a dorsal region resembling the normal pituitary tissue of the anterior lobe (al) and a ventral part where YFP− and YFP+ cells are intermingled. The boundary between dorsal and ventral regions is indicated with a dashed line. At 20 weeks, the YFP− population contains abundant Ki67+ cells and has expanded dramatically (white asterisks) displacing the mostly quiescent YFP+ cells to the periphery. Around 35 weeks, mutant mice reach a humane end-point and tumours are mostly YFP− (asterisk). Note that some YFP+ cells are observed in the periphery (white arrow). Immunohistochemistry against synaptophysin shows that whilst the anterior lobe cells (al) are positive, tumours do not express this endocrine marker (black asterisk). Immunostaining against β-catenin showing the presence of cell groups (clusters, white arrows) with nucleo-cytoplasmic accumulation of β-catenin mostly at 18.5 dpc and 5 weeks of age. Subsequently, most of the tumour cells accumulate β-catenin (white asterisk). Results are representative of experiments conducted in at least three different mice for each stage. Scale bars: 100 μm
Exome sequencing of 15 murine tumours revealed the presence of a total of 272 mutations (average 18.13 mutations per tumour; Supplementary Data 2) of which 131 were in coding regions (average 8.73 mutations per tumour; Supplementary Data 2). Of a selection of 14 of these 131 mutations, we validated 12 by Sanger sequencing (i.e. they were identified in the same DNA used for exome sequencing) (Supplementary Data 2). Only three genes were recurrently mutated in either two or three separate tumours and no recurrent mutations were found present in all 15 tumours (Supplementary Data 2). A low mutational burden with few recurrent mutated genes, other than CTNNB1, has been described in human ACP33. To determine if these changes are representative of the mutagenic process occurring in human ACP, we carried out comparisons with the published exome sequence results of 12 human ACPs33. This revealed 12 genes carrying mutations in both human and mouse tumours, but none of these mutations were identical between species (Supplementary Data 2). Five of these 12 mutated genes harboured non-silent/intronic mutations in both species (Table 1). The functional significance of these mutations is currently unknown.
Together, these findings demonstrate that the activation of oncogenic β-catenin in Hesx1+ pituitary embryonic precursors leads to cell transformation and formation of somatic mutations-bearing tumours, which are mostly not derived from the Hesx1 cell lineage. Additionally, our time course analysis reveals that tumour initiation requires a latency period of several weeks and is preceded by formation of β-catenin-accumulating cell clusters in the pre-tumoural pituitary.
ACP clusters contain senescent cells with an activated SASP
The observation that cluster cells do not divide led us to hypothesise that the expression of mutant β-catenin may result in oncogene-induced senescence. We analysed the expression of a panel of senescence-associated markers by immunostaining in mutant mouse embryos at 18.5 dpc and 23 different human ACP samples to assess whether hallmark senescence features were present in cluster cells including: (i) cell cycle arrest; (ii) DNA damage and activation of a DDR and (iii) increased lysosomal content1, 38.
Cluster cells were negative for the mitotic marker phospho-histone H3 and did not incorporate 5-ethynyl-2′-deoxyuridine (EdU), a specific indicator of S phase. Cell death was not detected by caspase 3 immunostaining (Supplementary Fig. 2). Human β-catenin-accumulating cluster cells are also negative for Ki67 and caspase 336, 39. Murine and human clusters exhibited marked expression of p53 (TP53), p21 (CDKN1A) and p16 (CDKN2A), all crucial effectors of cell cycle arrest38 (Fig. 2a, b; Supplementary Fig. 3a, b). Cluster cells in mouse and human ACP expressed γ-H2AX, suggesting the presence of DNA damage40; as well as phospho-DNA-PKcs, PARP1 and phospho-ATM, therefore revealing DDR activation41, 42 (Fig. 2a, b; Supplementary Fig. 3c, d). The senescence-associated β-galactosidase (SA-βgal) staining is a classical assay to identify senescent cells by detecting the presence of the lysosomal enzyme β-d-galactosidase43, the product of the GLB1 gene44. SA-βgal staining on Hesx1Cre/+;Ctnnb1lox(ex3)/+ mutant pituitaries (Supplementary Fig. 3e) and double immunofluorescence for β-catenin and GLB1 revealed the expression of GLB1 in the cell clusters in both mouse and human ACP (Fig. 2a, b). LAMP1, LAMP2 and lysozyme C expression were also upregulated in cluster cells, suggesting the enlargement of the lysosomal compartment, another feature of senescent cells (Supplementary Fig. 3f, g)44, 45.

β-catenin-accumulating cluster cells in mouse and human ACP express senescence markers. Double immunostaining on histological sections of Hesx1Cre/+;Ctnnb1lox(ex3)/+ pre-tumoural pituitaries at 18.5 dpc (a) and human ACP tumours (b). Note that cluster cells (arrows) express lysosomal enzymes such as GLB1, encoding senescence-associated β-galactosidase, the cell cycle regulators p21 and p53, as well as the DNA damage and DNA damage response markers gamma histone 2AX and phospho-DNA-PKcs, respectively. Results are representative from experiments conducted in six to eight mice and 23 human ACP tumours. Normal brain was used as negative control. Scale bars: a 50 μm; b 100 μm
The activation of the NF-κB pathway is required for survival of senescent cells and activation of SASP11. As a read out of an activated NF-κB pathway, we assessed the presence of phospho-IκBα, as phosphorylation at Ser32 and Ser36 of IκBα is followed by proteasome-mediated degradation and pathway activation46. Mouse and human clusters expressed high levels of p-IκBα (Fig. 3a, b). RELA/p65 and NEMO, two crucial components of the NF-κB pathway, were broadly expressed throughout the tissue, including cluster cells of both species (Fig. 3a, b). Finally, to determine if senescence and the SASP response are initiated together with cluster formation and prior to any tumour initiation, we turned to the early stages of mutant pituitary development in the embryonic mouse model. Analysis of Hesx1Cre/+;Ctnnb1lox(ex3)/+ embryonic pituitaries from 10.5 to 18.5 dpc revealed a dynamic response for the different aspects of senescence in the cluster cells. Proliferation in β-catenin-accumulating cluster cells, as assessed by Ki67 and EdU incorporation, was higher at 10.5 dpc than 18.5 dpc, whilst expression of the cell cycle inhibitors p16 and p21 was detected in the majority of the cluster cells from 10.5 dpc (Supplementary Fig. 4a, b). The DDR and NF-κB pathways became active by 10.5–12.5 dpc (Supplementary Fig. 4c), whereas expansion of the lysosomal compartment occurred later in development around 16.5–18.5 dpc (Supplementary Fig. 4a, b, d). Therefore, the senescence/SASP response is initiated at early stages of development, soon after the emergence of tumours.

Cluster cells show activation of the NFκB pathway. Double immunostaining on histological sections of Hesx1Cre/+;Ctnnb1lox(ex3)/+ pre-tumoural pituitaries at 18.5 dpc (a) and human ACP tumours (b). Phospho-IκBα, a readout of activated NFκB pathway, as well as RELA and NEMO, two components of this pathway, are expressed in cluster cells (white arrows). Results are representative of experiments conducted in four to six mice. Scale bar: a 50 μm; b 100 μm
SASP activation results in alterations in normal tissue architecture and increased angiogenesis6, therefore, we sought to analyse the tumour microenvironment in Hesx1Cre/+;Ctnnb1lox(ex3)/+ pituitaries at 18.5 dpc. In controls, immunostaining against endomucin (EMCN), a sialoglycoprotein routinely used as a marker of endothelial cells47, revealed the presence of cells with a thin and elongated morphology surrounding the lumen of blood vessels in the anterior lobe of the control pituitaries (Fig. 4a). In contrast, Hesx1Cre/+;Ctnnb1lox(ex3)/+ pituitaries displayed a drastic expansion of the EMCN+ population, which formed large and dense groups of cells without the presence of a lumen (Fig. 4a). Double immunostaining against YFP and EMCN in the Hesx1Cre/+;Ctnnb1lox(ex3)/+;R26YFP/+ pituitaries revealed mutually exclusive staining with the EMCN cells occupying almost entirely the YFP− areas (Fig. 4b). Expression of extra-cellular matrix proteins such as fibronectin and laminin was altered in the Hesx1Cre/+;Ctnnb1lox(ex3)/+ pre-tumoural pituitaries when compared with controls (Fig. 4a). Interestingly, EMCN+ cells frequently formed ring-like structures surrounding YFP+ cells, which resembled the β-catenin-accumulating clusters. Triple immunostaining against SOX9, β-catenin and EMCN confirmed that the cells directly surrounding the clusters are SOX9+;EMCN+ (Fig. 4c). Together, these results suggest that cluster cells activate a senescence and SASP programme leading to changes in the cellular microenvironment of the pre-tumoural pituitary.

Tissue microenvironment changes in the pre-tumoural pituitary of Hesx1Cre/+; Ctnnb1lox(ex3)/+ mutants. a Immunostaining against endomucin (EMCN), fibronectin and laminin revealing changes in the parenchyma of the pre-tumoural pituitary at 18.5 dpc. Scale bar: 100 μm. b Mutually exclusive staining of YFP+ (detected with an anti-GFP antibody) and EMCN+ cells. Note the presence of YFP+ cells (arrows) surrounding a population of EMCN+ cells. Scale bar: 100 μm. c Triple immunostaining showing the co-expression of EMCN and SOX9 in large group of cells around the β-catenin-accumulating clusters (arrows). Results are representative of experiments conducted in six mice. Scale bar: 100 μm
ACP clusters show a senescence/SASP molecular signature
The expression of oncogenic β-catenin in Sox2+ pituitary stem cells in Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ mice at 4–6 weeks of age results in cluster formation and induction of paracrine tumours28. However, the molecular signature of the cluster cells in Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ mice has not yet been revealed. To this end, we first induced Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ mice at 4 weeks of age by tamoxifen administration and analysed the pituitaries 4 weeks later. Following dissociation into single-cell suspensions, YFP+ and YFP− cells were isolated by flow sorting and subjected to qRT-PCR analysis. This revealed a gene expression profile consistent with the presence of β-catenin-accumulating cluster cells in the YFP+ population (Supplementary Fig. 5b).
Next we sought to reveal the molecular signature of cluster cells relative to normal Sox2+ cells not expressing oncogenic β-catenin. Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ mutant and Sox2CreERT2/+;R26YFP/+ control mice were tamoxifen induced at 4 weeks of age and pituitaries dissociated and subjected to RNA sequencing 4 weeks later. The expression data for all of the 27,179 genes are included in Supplementary Data 3 and the demonstration of a successful experimental approach in Supplementary Fig. 5a. Of note, differential expression studies demonstrated the upregulation of several members of the hedgehog (HH), fibroblast growth factor (FGF), bone morphogenetic protein (BMP), wingless-related integration site (WNT) and transforming growth factor β (TGFβ) families as well as CXC chemokines and interleukins (Supplementary Data 3). These results support the notion that cluster cells are signalling hubs in the pre-tumoural pituitary.
To assess in an unbiased manner whether cluster cells in Sox2CreERT2/+;Ctnnb1lox(ex3)/+ and Hesx1Cre/+;Ctnnb1lox(ex3)/+(previously characterised48) mice activate senescence and a SASP, we specifically compared the expression profiles of the murine clusters with established molecular signatures for OIS and SASP obtained from human IMR90 ER:RAS fibroblasts. These cells express a Steroid Receptor-KRASG12V chimaeric fusion protein that triggers growth arrest, senescence and SASP13. Gene set enrichment analysis (GSEA) showed that the gene expression profiles of the clusters in both mouse models were significantly enriched for OIS and SASP genes (Fig. 5a, c). This was further validated by qRT-PCR (Fig. 5b, d). SASP activation was further investigated using semi-quantitative ELISA cytokine arrays, which demonstrated the upregulation of several SASP-related proteins, such as ICAM1, bFGF, IL1A, IFNG, OPN, IL6, IGFBL6, MMP3, MMP2, RNAKL1, CXCL1, CXCL11, IL7, TIMP2, VEGFR1 and CCL11) (Fig. 5e).

Mouse and human β-catenin-accumulating clusters share a common signature of senescence and SASP. a–d Gene set enrichment analysis (GSEA) (a, c) revealing that clusters from both mouse models, the Hesx1Cre/+;Ctnnb1lox(ex3)/+ and Sox2CrERT2e/+;Ctnnb1lox(ex3)/+;R26YFP/+, show a molecular signature of oncogene-induced senescence (OIS) and senescence-associated secretory phenotype (SASP). qRT-PCR analysis showing the upregulation of several senescence and SASP factors in both mouse models (b, d). Bars in b and d represent the mean, and error bars represent standard error of the mean (SEM) of three biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001, Student’s t test. e Several cytokines are upregulated in the Hesx1Cre/+;Ctnnb1lox(ex3)/+ mutant relative to the Hesx1+/+;Ctnnb1lox(ex3)/+ control pituitaries as measured in a mouse cytokine antibody array. f Gene set enrichment analysis (GSEA) demonstrating that laser-capture micro-dissected human clusters show a molecular signature of oncogene-induced senescence (OIS) and senescence-associated secretory phenotype (SASP). g Hierarchical clustering analysis of RNA-data sets from cluster and non-cluster cell populations indicating coherent grouping between specific cell compartments. h, i GSEA of the top 100 or 500 most upregulated genes in human clusters and the whole transcriptome of mouse clusters from both models demonstrating a common molecular signature
To determine whether these findings were relevant to human ACP, we assessed the degree of molecular equivalence between mouse and human ACP clusters. We recently analysed the molecular signatures of specific tumour cell compartments in human ACP including the β-catenin-accumulating cell clusters, by combining laser-capture microdissection and RNA-Sequencing (J.R. Apps and J.P. Martinez-Barbera, manuscript submitted, Array Express accession code: E-MTAB-5266). Hierarchical clustering analysis revealed the grouping of Hesx1Cre/+;Ctnnb1lox(ex3)/+, Sox2CreERT2/+;Ctnnb1lox(ex3)/+ and human clusters, suggesting similar molecular profiles (Fig. 5g), while GSEA confirmed that human clusters also have a molecular signature of senescence and SASP (Fig. 5f). Moreover, GSEA analysis of Hesx1Cre/+;Ctnnb1lox(ex3)/+ and Sox2CreERT2/+;Ctnnb1lox(ex3)/+ cluster gene expression profiles showed a significant enrichment for the top 100 and 500 most upregulated genes in human clusters (Fig. 5h, i). Together, these analyses support that human and mouse clusters are equivalent structures, sharing a common signature of senescence and SASP.
Aged Sox2+ stem cells show reduced tumour-inducing potential
We sought to determine whether the age at which Sox2CreERT2/+;Ctnnb1lox(ex3)/+ mice were targeted to express the oncogenic β-catenin may have an effect on paracrine tumourigenesis.
Sox2CreERT2/+;Ctnnb1lox(ex3)/+ mutant mice were tamoxifen induced at either 1 month, when pituitary stem cells are still actively supporting organ growth, or at 6–9 months of age when contribution from Sox2+ stem cells to homeostasis is low. Pituitaries were analysed at 3 months post induction and assessed for tumour formation. Histological analysis revealed that 15/15 mice that were induced at 1 month of age developed pituitary tumours with a total of 48 individual lesions (average 2–4 tumours per pituitary). However, only 3 out of 17 mice induced at 6–9 months of age developed pituitary tumours with only five individual lesions observed (p < 0.0001; Table 2).
We decided to explore whether the reduced tumour burden in older Sox2CreERT2/+;Ctnnb1lox(ex3)/+ mice may be related to an attenuated senescence and SASP response in the β-catenin-accumulating clusters. To achieve this, YFP+ cells were isolated by flow cytometry from Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ mutant and Sox2CreERT2/+;R26YFP/+ control mice that were induced at either one month or 6–9 months of age (Fig. 6a). Relative to age-matched control YFP+ cells, YFP+ cells from older mice activated the WNT pathway and a senescence/SASP programme (Fig. 6d). Although both age groups contained Ki67−/p21+ β-catenin-accumulating clusters (Fig. 6b), the average number of cells in the clusters of young-induced Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ mutant mice was 3.16 ± 1.57 vs. 1.95 ± 1.12 in older-induced animals, affirming that clusters from older mice were smaller in size (p < 0.0001, Student’s t test; 57 clusters counted in three young Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ pituitaries and 94 clusters counted in six older Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ pituitaries). In addition, increased Ki67 index was observed only in non-cluster cells in the younger, but not in the older Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ pituitaries, suggesting that Sox2+ cluster cells from younger mice can induce a proliferative response in surrounding cells (Fig. 6c).

Aged Sox2+ stem cells generate β-catenin-accumulating cell clusters with reduced senescence and SASP responses. a Schematic diagram of the experimental designs. Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ and Sox2CreERT2/+;R26YFP/+ mice were tamoxifen induced at either 1 or 6 month of age and the anterior pituitary collected 4 weeks post induction and processed for histological and molecular analysis of YFP+ cells isolated by FACS (n = 8 mice per group). b Double immunostaining on histological sections of Sox2CreERT2/+; Ctnnb1lox(ex3)/+; R26YFP/+ pituitaries revealing the presence of β-catenin-accumulating cell clusters positive for p21, but not for Ki67. Scale bar: 100 µm. c Quantitative analysis showing a significant higher Ki67 proliferation index in non-cluster cells in Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ tumoural relative to Sox2CreERT2/+;R26YFP/+ control pituitaries in tamoxifen-induced mice at 1 month (black asterisk), but not at 6 months. Increased proliferation index is also observed in non-cluster cells in Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ pituitaries of mice induced at 1 month relative to those induced at 6 months (red asterisk). d qRT-PCR analysis showing the upregulation of the WNT pathway target genes Axin2 and Lef1, as well as of several senescence and SASP factors in the Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ mutant pituitaries relative to the Sox2CreERT2/+; R26YFP/+ controls. e qRT analysis showing the relative expression of several senescence and SASP factors in Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ mice tamoxifen induced at either 1 (young) or 6 (older) months of age. The expression of several senescent and SASP factors is significantly reduced in the older compared with the young mice. Bars in c, d and e represent the mean, and error bars represent standard error of the mean (SEM) of three biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001, Student’s t test
Importantly, comparative analysis between the YFP+ fractions of Sox2CreERT2/+;Ctnnb1lox(ex3)/+;R26YFP/+ mutants revealed a significant reduction in the expression of senescence and SASP factors in mice induced at 6–9 months relative to the one month-induced mice (Fig. 6e). We also observed an age-dependent increase in the expression of senescence-associated markers, such as p16 (Cdkn2a) and p53 (Trp53), in non-mutant Sox2+ cells in Sox2CreERT2/+;R26YFP/+ control pituitaries, which may underlie the ameliorated senescence/SASP response of older relative to younger mutant Sox2+ stem cells (Supplementary Fig. 6). Together, these analyses demonstrate that expression of oncogenic β-catenin in Sox2+ stem cells in 6–9 months old mice is less tumourigenic and leads to weakened senescence and SASP responses with formation of smaller β-catenin-accumulating clusters.
Apc-deficient clusters lack tumour-inducing potential
The data previously described in Hesx1Cre/+;Ctnnb1lox(ex3)/+ and Sox2CreERT2/+;Ctnnb1lox(ex3)/+ mice are compatible with a model whereby senescent cells in the β-catenin-accumulating clusters of the pre-tumoural pituitary may initiate tumourigenesis by activating a robust SASP programme. Therefore, we sought to explore whether the reduction or ablation of senescent cells may prevent or delay tumourigenesis.
The senolytic compounds ABT-263 and ABT-737 have been shown to selectively ablate senescent cells in vitro and in vivo49, 50. In agreement with these findings, ex vivo culture of 18.5 dpc Hesx1Cre/+;Ctnnb1lox(ex3)/+ pre-tumoural pituitaries in the presence of ABT-263 and ABT-737 revealed a reduction in the average size of the clusters, but this did not reach significance (number of cells per cluster: vehicle 2.46 ± 1.26, 41 clusters counted from three pituitaries; ABT-263, 1.4 ± 0.49, p = 0.47, 40 clusters counted from four pituitaries; ABT-737, 1.8 ± 0.62, p = 0.70, 38 clusters counted from three pituitaries; Student’s t test) (Supplementary Fig. 7c, d). In contrast, treatment of IMR90 ER:RAS senescent fibroblasts with these two senolytic agents significantly increased cell death relative to control non-senescent cells (Supplementary Fig. 7a, b). The distinct target cell type (i.e. epithelial cluster cell rather than fibroblast) may confer a resistance to these drugs and, additionally, drug delivery and dose may require further optimisation. The drugs target genes Bcl2, Bcl-xl and Bcl-w are all expressed in the cluster cells, but not upregulated.
Since chemical ablation of senescent cells was not strong enough to reduce significantly the size of the clusters, we sought to explore whether a genetic approach may impair the senescent/SASP response more robustly. It has been shown that the strength of the senescence/SASP response depends on the oncogenic hit; e.g. the activation of the mitogen-activated protein kinase (MAPK) pathway by BRAFV600E is a more potent senescence inducer than KRASG12V 51. We reasoned that the deletion of tumour suppressor Apc in Hesx1Cre/+;Apcfl/fl mice would lead to the activation of the WNT pathway in embryonic precursors and the accumulation of wild-type β-catenin rather than its degradation-resistant form, which is encoded by the oncogenic Ctnnb1 locus in Hesx1Cre/+;Ctnnb1lox(ex3)/+ mice. We expected wild-type β-catenin to be a weaker senescence inducer than oncogenic β-catenin, resulting in reduced senescence/SASP activation.
Hesx1Cre/+;Apcfl/fl mice were born at the expected Mendelian ratios, were normal and fertile with proper specification of pituitary endocrine lineages (Supplementary Data 4; Supplementary Fig. 8a). Although slight pituitary hyperplasia was consistently observed, no pituitary tumours ever formed in Hesx1Cre/+;Apcfl/fl mice (n = 61), even when followed until 18 months of age (n = 17) (Fig. 7a; Supplementary Fig. 8a). Immunostaining studies revealed the presence of nucleo-cytoplasmic β-catenin-accumulating clusters in Hesx1Cre/+;Apcfl/fl embryos. These clusters resembled their oncogenic β-catenin counterparts in many ways, such as in the expression of SOX2, but not SOX9, and WNT pathway activation as evidenced by the over-expression of the WNT-target gene Axin2 (Fig. 7d; Supplementary Fig. 8b). Additionally, cluster cells in the Apc mutant pituitary were negative for Ki67, phospho-Histone H3, or Caspase 3 and expressed the cell cycle inhibitors p21 and p16 (Fig. 7b; Supplementary Fig. 8c, d). Moreover, these clusters were strongly positive for DDR and NF-κB activation markers such as phospho-DNA-PKcs, PARP-1 and phospho-IκBα, indicative of the activation of a senescence programme (Fig. 7b; Supplementary Fig. 8d). Despite the similarities in gene expression, the average cluster size at 18.5 dpc in Hesx1Cre/+;Apcfl/fl pituitaries was 1.97 ± 1.36 cells, rather than 5.89 ± 0.33 in Hesx1Cre/+;Ctnnb1lox(ex3)/+ embryos (p < 0.0001, Student’s t test; 126 clusters counted for Hesx1Cre/+;Ctnnb1lox(ex3)/+ and 108 clusters counted for Hesx1Cre/+;Apcfl/fl in three different pituitaries).

Deletion of the tumour suppressor Apc results in β-catenin-accumulating senescent clusters with reduced SASP. a Brightfield and haematoxylin–eosin pictures of wild-type and Hesx1Cre/+;Apcfl/fl pituitaries, and Hesx1Cre/+;Ctnnb1lox(ex3)/+ pituitary tumours. Note that the Apc-deficient pituitary is enlarged but no tumours are apparent and it is histologically normal. The age of the mice is indicated in weeks. Scale bar: 1 mm. b Double immunostaining revealing the expression of p21, phospho-IκBα and phospho-DNA-PKcs in the β-catenin-accumulating clusters, indicating cell cycle inhibition and activation of the NFκB and DNA damage response pathways, in both Hesx1Cre/+;Apcfl/fl and Hesx1Cre/+;Ctnnb1lox(ex3)/+ pituitaries at 18.5 dpc. Note that the clusters are smaller in the Hesx1Cre/+;Apcfl/fl compared with the Hesx1Cre/+;Ctnnb1lox(ex3)/+ pituitaries. Scale bar: 50 μm. c qRT-PCR analysis showing the relative expression of several genes in Hesx1Cre/+;Apcfl/fl relative to Hesx1Cre/+; Ctnnb1lox(ex3)/+ pituitaries at 18.5 dpc. The expression of the WNT pathway targets Axin2 and Lef1 are similar between the two genotypes, but several senescence and SASP factors show reduced expression levels in the Hesx1Cre/+;Apcfl/fl compared with the Hesx1Cre/+;Ctnnb1lox(ex3)/+ pituitaries. Bars represent the mean, and error bars represent standard error of the mean (SEM) of three biological replicates. *p < 0.05, **p < 0.01, Student’s t test. d In situ hybridisation on histological sections of embryos at 14.5 dpc showing the expression of Axin2 in small cell clusters and very reduced or absent detection of Bmp4, Fgf3 and Shh in the Apc-deficient pituitaries. Scale bar: 100 μm. e Triple immunostaining revealing the expression of β-catenin, SOX9 and EMCN. Note that the large areas of EMCN+;SOX9+ cells surrounding the β-catenin-accumulating clusters are not present in the Hesx1Cre/+;Apcfl/fl pituitaries. Scale bar: 50 μm. f Quantification of the EMCN+ areas demonstrating that wild-type and Apc-deficient pituitaries show similar levels, which are significantly increased in the Hesx1Cre/+;Ctnnb1lox(ex3)/+ pituitaries. Bars represent the mean, and error bars represent standard error of the mean (SEM) of three biological replicates. ***p < 0.001, one-way ANOVA. All results are representative of at least three biological replicates
qRT-PCR analysis comparing Hesx1Cre/+;Apcfl/fl to Apcfl/+ control pituitaries revealed no significant differences in the expression levels of several senescence and SASP factors including Cdkn1a, Cdkn2a, Shh, Bmp4, Il1a, Il1b, Il6, Cxcl1 and Tgfb1 (Supplementary Fig. 8e). However when comparing expression levels of senescence/SASP factors relative to Hesx1Cre/+;Ctnnb1lox(ex3)/+ pituitaries, these were reduced in Hesx1Cre/+;Apcfl/fl mutants (Fig. 7c), suggesting an overall reduction of the senescence/SASP response upon Apc deletion and in line with the formation of smaller clusters. In situ hybridisation previously confirmed the upregulation of Axin2, Bmp4, Fgf3 and Shh in Hesx1Cre/+;Ctnnb1lox(ex3)/+ clusters27. In contrast, despite Hesx1Cre/+;Apcfl/fl clusters expressing Axin2 and demonstrating WNT pathway activation, the expression of Bmp4, Fgf3 and Shh expression was not detectable (Fig. 7d). Moreover, the abundant SOX9+;EMCN+ cells surrounding the clusters in the Hesx1Cre/+;Ctnnb1lox(ex3)/+ pituitaries (Fig. 4c) were not apparent in Hesx1Cre/+;Apcfl/fl mutant glands (Fig. 7e), and overall quantification of the area covered by endomucin positive cells revealed values comparable to control littermates (Fig. 7f). In a Hesx1Cre/+;Apcfl/fl pituitary where a larger cluster was identified, accumulation of EMCN+ cells was observed around it (Supplementary Fig. 8f), suggesting that a threshold of SASP factors may be needed to elicit the changes in tissue architecture. Finally, no tumours developed in Sox2CreERT2/+;Apcfl/fl mice induced from 4 to 8 weeks of age in a follow up of up to a year (n = 7 mice). Together, these studies demonstrate that Apc deletion in pituitary embryonic precursors/stem cells leads to reduced senescence/SASP response and is not tumorigenic.
Discussion
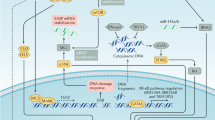
In this paper, we show that the expression of oncogenic β-catenin and activation of the WNT pathway in Hesx1+ embryonic pituitary precursors or Sox2+ pituitary stem cells in young mice results in the activation of a robust SASP in targeted cells and the subsequent paracrine formation of tumours. These mouse tumours carry a low burden of somatic mutations that are mostly non-recurrent, which overlap with human ACP. To our knowledge, this is the first time that SASP activation is associated with non-cell autonomous cell transformation (i.e. induction of somatic mutations) and tumour initiation in an in vivo murine model of a human tumour. Of note, we refer to cell transformation to indicate the occurrence of somatic mutations not present in the germline and the acquisition of a proliferative advantage of the tumour relative to the non-tumour cells. However, mouse and human ACP do not harbour mutations in critical cancer genes (e.g. p53, p16, Myc, Braf, Egfr among others) suggesting that these tumours are not fully transformed. This is not surprising as human ACP is a benign tumour that very rarely progresses into a malignancy. A summary of the main findings of this study is shown in Fig. 8.

Schematic summary of the main findings of this research. a Targeting Sox2+ stem cells from young adults, or Hesx1+ embryonic precursors to express oncogenic β-catenin, results in formation of senescent cell clusters and a robust SASP. This leads to cell transformation and generation of the cell-of-origin of the paracrine tumours, thus the tumours are not derived from the mutation-sustaining cells. b In contrast, the expression of oncogenic β-catenin in Sox2+ stem cells of older mice leads to a reduced tumour incidence or the failure to produce tumours. In addition, upregulation of the WNT pathway through the deletion of Apc in young adult stem cells or embryonic precursors also fails to induce tumours. In all cases described in b, there is attenuated senescence and SASP activation
In the embryonic mouse ACP model, tumour progression does not start immediately after birth, but only after a latency period in which the pre-tumoural pituitary contains senescent clusters with an activated SASP. This non-linear growth dynamics have recently been characterised by magnetic resonance imaging37. The pre-tumoural pituitary also shows abnormal tissue architecture and overall slow cell proliferation. However, once rapidly dividing tumour cells appear, the clusters and most of the normal pituitary tissue are displaced. This growth pattern is compatible with the clusters’ activities being required to initiate a process eventually leading to tumour initiation (hit-and-run model), rather than promoting tumour progression, which may be self-sustaining. Recently, it has been shown that senescence cells through SASP can induce dedifferentiation and stem cell marker expression52. Transcriptomics failed to reveal the expression of Hesx1, but stem cell markers such as SOX9, Klf2, Klf4 and Cmyc were upregulated in mouse tumours (J.R. Apps and J.P. Martinez-Barbera, manuscript submitted), sometimes in close proximity to the senescent cluster cells (i.e. SOX9; Fig. 4c)27. Immunostaining against SOX2 showed lack of expression of this stem cell marker in the murine tumours (Supplementary Fig. 9). In human ACP, the expression of stem cell markers (e.g. SOX2, SOX9, KLF4 and OCT4) has been found to be upregulated53. These results are compatible with a potential role of the senescent cluster cells in the promotion of reprogramming in vivo, a hypothesis that merits further research.
Elegant genetic experiments have shown that senescence is important to prevent tumourigenesis in mouse models of pituitary adenoma54. In agreement with this notion, our data suggest that senescence has an early cell-autonomous function that prevents the expansion of cluster cells harbouring oncogenic β-catenin. However, we reveal an additional non-cell autonomous role of senescent cells, mediated by the persistent activation of SASP resulting in paracrine cell transformation and tumourigenesis. Ablation of the senescent cluster cells at specific time points using chemical or genetic approaches will further help dissect their role in tumourigenesis. In particular, it will help determine when and for how long their activities are required for tumour initiation and/or maintenance.
We also reveal molecular similarities between the mouse and human clusters, which provide insights into the pathogenesis of human ACP. In contrast to the mouse tumours, the β-catenin-accumulating clusters are present in fully developed human ACP and tend to be located at the tips of finger-like epithelial protrusions that invade the brain36, 37. Analysis of a human ACP xenograft model suggest that the activities of the clusters are important in orchestrating cell migration and tumour invasion36. We show that β-catenin clusters undergo senescence and initiate SASP during embryonic pituitary development in the mouse, and this supports the notion that childhood-onset ACP is a developmental tumour. This proposition is substantiated by the scrutiny of clinical data, which has revealed that paediatric ACP patients suffer significant height deficits around 12 months of age, many years before ACP diagnosis, suggesting potential growth hormone deficiency55. Of note, somatotrophs, which express growth hormone, are clearly defective in the Hesx1Cre/+;Ctnnb1lox(ex3)/+ ACP model by the end of gestation27. Therefore, clusters may develop embryonically in human patients and be present in the pituitary gland of the newborn prior to the appearance of any tumour mass. In fact, foetal ACPs displaying nucleo-cytoplasmic β-catenin-accumulating cells have recently been diagnosed in human foetus at 18 weeks56. Additionally, we show that the SASP response is attenuated with age in Sox2+ pituitary stem cells in older mice, which is in agreement with ACP being primarily a paediatric tumour29.
Paracrine tumourigenesis has been reported in other tumours and cancers, but so far not associated with senescence and SASP. Over-expression of platelet-derived growth factor (PDGF) (also a SASP factor) in neural precursors in mice and rats is sufficient to recruit normal neural stem and progenitor cells that acquired genetic aberrations and become malignant, capable of generating tumours upon transplantation57,58,59. Likewise, conditional deletion of Notch1 in mouse skin epidermis60 or activation of β-catenin in hair follicles stem cells by deleting Ctnnb1 exon 361 leads to over-activation of the WNT/β-catenin pathway and induction of tumours/growths in a non-cell autonomous manner. Additionally, the expression of a constitutively active form of MEK1, activating the ERK MAPK pathway, in mouse epidermis results in skin polyps, which are mostly formed by cells not carrying oncogenic MEK162. More strikingly, in a mouse model for leukaemia the expression oncogenic β-catenin (i.e. Ctnnb1 deletion of exon3) in osteoblast precursors is sufficient to generate oncogenic hematopoietic stem cells capable of giving rise and propagating acute myeloid leukaemia63. Whether in these models the tumour-inducing cells are senescent and have activated SASP is an important aspect that merits further research. Our results support the possibility that the activities of the senescent cells result in changes in the cellular microenvironment driving genetic and possibly, epigenetic changes in neighbouring cells, which may fuel tumour initiation in some of these neoplasias.
In summary, this study provides evidence for a mechanism by which embryonic progenitors and postnatal stem cells can contribute to tumourigenesis in a non-cell autonomous manner, identifying senescence and SASP as mediators of this tumour-inducing potential. Recent research has demonstrated that clearance of senescent cells, either naturally occurring in aged mice or diseased related, extends lifespan, improves the physiological function of several organs and delays tumourigenesis49, 50, 64. Although the ablation of the cluster cells in the ACP mouse model was only partial, these results reinforce the idea that cluster cells are senescent and suggest that senolytic compounds may be of potential therapeutic interest against human ACP. The development of more specific senolytics capable of targeting a variety of senescent cells, including pituitary epithelial cells targeted with oncogenic β-catenin, will be required to further study the potential anti-tumourigenic effects of senescence ablation in the context of ACP and other tumours. In addition, further research is required to identify the pro- and anti-tumourigenic mediators in the SASP, and the mouse models presented here are suitable tools to dissect these opposite functions.
Methods
Mice and human samples of ACP
The Ctnnb1lox(ex3)/+, Sox2CreERT2/+, Hesx1Cre/+, ROSA26flox-stop-YFP/+, Apcflox/+ and ROSAmTmG/+ mice have been previously described28, 48, 65, 66. Sox2CreERT2/+; Ctnnb1lox(ex3)/+; ROSA26flox-stop-YFP/+ and Sox2CreERT2/+; ROSA26flox-stop-YFP/+ mice were induced with a single intra-peritoneal injection of tamoxifen (Sigma, T5648) at a dose of 0.15 mg per gram of body weight. All mouse procedures were performed under licence, following UK Home Office Animals (Scientific Procedures) Act 1986 and local institutional guidelines (UCL ethical review committee). A humane end-point was chosen to avoid severe adverse effects and minimise suffering of mice harbouring tumours, according to UK Home Office and local Ethical Committee regulations. Samples of human ACP were obtained from the Department of Histopathology at Great Ormond Street Hospital for Children and the CCLG tissue bank with full consent (Research Ethics Committee application, REC 14 LO 2265).
Flow-activated cell sorting
Anterior pituitaries from age- and sex-matched mice were dissociated mechanically into single-cell suspensions following incubation in enzyme mix (0.5% w/v Collagenase type 2 (Lorne Laboratories Ltd.), 0.1× Trypsin (Gibco) and 50 μg/ml DNaseI (Worthington) with 2.5 μg/ml Fungizone (Gibco) in Hank’s Balanced Salt Solution (HBSS (Gibco) for 4 h at 37 °C. Cells were then washed in HBSS and suspended in PBS supplemented with 1% Foetal Calf Serum (PAA), 25 mM HEPES and 1 ng/μl of propidium iodide (PI, BD Pharmigen). Cells were flow sorted for YFP and PI fluorescence using a MoFlo XPD (Beckman Coulter, Fullerton, CA, USA) Flow Cytometer. Fluorescence was detected using a 530/540 filter, where PI positive cells were excluded and the YFP positive and negative fractions collected directly into RLT buffer (Qiagen) supplemented with 1% 2-mercaptoethanol (Sigma) for subsequent RNA isolation.
Expression analyses in histological sections
Whole-mount X-gal staining, immunostaining and in situ hybridisation on 4–6 µm paraffin sections were performed as previously described27. At least three pituitaries or tumours and between two to six non-consecutive slides were analysed. As negative controls, sections were hybridised with sense riboprobes for in situ hybridisation or secondary antibody alone for immunohistochemistry/immunofluorescence. A list of the antibodies and the antigen retrieval methods used is presented in Supplementary Data 1.
EdU labelling
Pregnant females were injected intra-peritoneally with 100 mg/kg of EdU and embryos dissected 90 min later. After histological processing and sectioning (see above), double immunostaining for β-catenin and EdU was conducted using the Click-It EdU imaging kit (Invitrogen) according to the manufacturer’s instructions.
RNA extraction and cDNA generation
Flow sorted cells from Sox2CreERT2/+;Ctnnb1lox(ex3)/+;ROSA26YFP/+ and Sox2CreERT2/+;ROSA26YFP/+ mice were processed for total RNA extraction using the RNeasy Micro kit (Qiagen). In total, 5 μl of eluted RNA was then amplified using the Ovation Pico WTA V2 kit (NuGEN), followed by purification of the resultant cDNA using the QIAquick PCR purification kit (Qiagen). Pituitaries from Hesx1Cre/+;Ctnnb1lox(ex3)/+ and control littermates were dissected at 18.5 dpc. The posterior lobe of the pituitary was discarded and the intermediate and anterior lobes were processed for total RNA extraction using the RNeasy Micro kit (Qiagen). Approximately 1 μg of total RNA was reverse transcribed to cDNA using the Transcriptor First Strand cDNA Synthesis Kit and random hexamers (Roche).
Quantitative real-time PCR and RNA-seq
Quantitative real-time PCR reactions were run in triplicate using the iTaq SYBR Green (BIORAD) and replicated using a minimum of four independent samples for each genotype. Primer sequences are included in Supplementary Data 5. For Shh, we used the Qiagen QuantiTect Primer Assay (Cat. no. 249900). Results were analysed using the ΔΔCt method. For RNA-Seq of Sox2CreERT2/+;Ctnnb1lox(ex3)/+;ROSA26YFP/+ and Sox2CreERT2/+;ROSA26YFP/+ flow-sorted YFP+ cells, isolated RNA was processed by the SMARTer V4 low input assay kit (Clontech) to generate amplified cDNA using the strand-switching protocol. Library preparation was performed with 200 pg of amplified cDNA using the Nextera XT preparation kit with 12 cycles of PCR. Sequencing was then performed on an Illumina NextSeq 500 with 43 bp paired end reads. Sequencing data was then aligned in BaseSpace. FASTQ and BAM files are available on Array Express (E-MTAB-5538), Differential expression was performed using DESeq267. For GSEA, genes were ranked by the Wald statistic and GSEA performed using the pre-ranked tool in GSEA v2.2 (Broad Institute)68. The WNT pathway gene set was downloaded from the hallmark molecular signatures database v5.2 (Broad Institute)68.
Analysis of the relationship between murine and human ACP
To explore the relationship between the ACP models and human ACP, gene sets of the 100 and 500 most upregulated genes in human ACP were generated and GSEA performed as above. In addition, expression data (normalised counts or intensity values) of 11,347 genes, for which expression data was available between non-cluster and cluster cells from human ACP (array express accession no: E-MTAB-5266), Sox2CreERT2/+;Ctnnb1lox(ex3);R26YFP/+ tamoxifen-induced mice (E-MTAB-5538) and Hesx1Cre/+;Ctnnb1lox(ex3)/+ (E-MEXP3492) animals were median scaled and merged. Clustering was performed using Spearman’s correlation and Ward’s algorithm.
Exome sequencing of mouse tumours
Fully developed pituitary tumours and matched tail samples of 15 Hesx1Cre/+;Ctnnb1lox(ex3)/+ mice were collected at humane end-points. DNA was extracted using the DNeasy bloods and tissue kit (Qiagen) and exonic DNA was captured using the Agilent whole exome capture kit (SureSelect Mouse All Exon). Captured material was indexed and sequenced on the Illumina platform at the Wellcome Trust Sanger Institute at 30× depth. Raw pair end sequencing reads were aligned with BWA-mem to the GRCm38 mouse reference genome. Duplicated reads were marked using biobambam. Somatic variants were detected using CaVEMan, an expectation maximisation-based somatic substitution-detection algorithm. Detected somatic variants were then filtered using an array of quality filters and common mouse genome variants were excluded69, 70. A sub-selection of 14 genes, where mutant variant allele frequencies were greater than 20% and at least three mutant reads were detected, were validated by Sanger sequencing. Primer were designed in flanking regions (Supplementary Data 2). Sequencing of the PCR fragments was performed by Source BioScience (UK).
Semi-quantitative ELISA cytokine arrays
Mouse C1000 cytokine arrays (AAM-CYT-1000-4, RayBiotech) were used to quantify the expression of SASP factors in protein extracts from whole pituitaries dissected at 18.5 dpc according to manufacturer’s instructions (sex was not determined in the embryos). For each experimental sample, total protein was extracted from four pituitaries using the cell lysis solution provided in combination with a TissueRuptor (Qiagen) and passed through a Qiashredder column (Qiagen). Total protein was then measured using BradfordUltra reagent (Expedeon). ELISA membranes were blocked for 30 min using blocking buffer provided by the manufacturer and then incubated overnight at 40 °C with 70 μg of total protein. Following extensive washes, membranes were incubated with biotinylated antibodies and developed according to manufacturer’s indications. Chemiluminescence detection was conducted with a ChemiDoc gel documentation system (BioRad) and imaged using ImageLab Software (Version 5.2.1, BioRad). Analysis of high-resolution images from each membrane was conducted using Fiji/ImageJ and the Protein Microarray Analysis macro (IRB, Barcelona). Calculation of fold changes in expression was conducted according to manufacturer’s instructions.
Assessment of clonogenic potential
Anterior pituitaries were dissected and enzyme treated as described for flow-activated cell sorting, above. This was followed by mechanically dissociation into a single-cell suspension and subsequent washing in Hank’s Balanced Salt Solution. Cells were placed directly into Adherent Pituitary Stem Cell medium (DMEM-F12; Gibco) containing 5% Foetal Calf Serum (PAA), 20 ng/ml bFGF (R&D Systems) and 50 ng/ml cholera toxin (Sigma). Cells were plated into six-well plates at a density of 1000, 2000 and 4000 cells per well for drug or vehicle treated. After seven days in culture, adherent colonies were fixed, washed with PBS and stained using Meyer’s Haematoxylin (Sigma) for 10 min and counted manually.
Ex vivo culture of mouse pituitaries and ABT-737 treatment
For ex vivo pituitary culture, anterior pituitaries were isolated from Hesx1Cre/+;Ctnnb1lox(ex3)/+ embryos at 18.5 dpc, placed on top of 0.2 µM Whatman filters (SLS) in 24 well plates containing 500 µl of media (DMEM-F12 (Gibco), 1% Pen/Strep (Sigma) and 1% FBS (PAA) supplemented with either ABT-737 at 2.5 µM (Adooq Bioscience), ABT-263 at 1 µM (Caymen Chemical) or vehicle. Anterior pituitaries were cultured for 3–4 days, changing media every day and subsequently processed for histological analysis.
Statistical analyses
Results are presented as means ± s.e.m. unless otherwise indicated. One-tailed Student’s t test was performed to compare the means of two groups. Significance was assumed at P < 0.05. Sample size varied, but a minimum of three biological replicas were used for analysis. Statistical analyses were performed using GraphPad software. No samples were excluded from analyses. No formal blinding or randomisation was used.
Code availability
Codes are available upon request
References
Campisi, J. Aging cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705 (2013).
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. & Lowe, S. W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 (1997).
Alcorta, D. A. et al. Involvement of the cyclin-dependent kinase inhibitorp16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl Acad. Sci. USA 93, 13742–13747 (1996).
Dhomen, N. et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell 15, 294–303 (2009).
Di Micco, R. et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642 (2006).
Coppe, J. P., Desprez, P. Y., Krtolica, A. & Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118 (2010).
Maciel-Baron, L. A. et al. Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli. Age (Dordr.) 38, 26 (2016).
Kipling, D. et al. A transcriptomic analysis of the EK1.Br strain of human fibroblastoid keratocytes: the effects of growth, quiescence and senescence. Exp. Eye Res. 88, 277–285 (2009).
Coppe, J. P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 (2008).
Rodier, F. et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 (2009).
Chien, Y. et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 25, 2125–2136 (2011).
Kuilman, T. et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 (2008).
Acosta, J. C. et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990 (2013).
Tamm, I., Kikuchi, T., Cardinale, I. & Krueger, J. G. Cell-adhesion-disrupting action of interleukin 6 in human ductal breast carcinoma cells. Proc. Natl Acad. Sci. USA 91, 3329–3333 (1994).
Tammela, T. et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature https://doi.org/10.1038/nature22334 (2017).
Kang, C. et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612 (2015).
Liu, D. & Hornsby, P. J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 67, 3117–3126 (2007).
Sparmann, A. & Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 6, 447–458 (2004).
Kang, T. W. et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551 (2011).
Yoshimoto, S. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101 (2013).
Bavik, C. et al. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 66, 794–802 (2006).
Krtolica, A., Parrinello, S., Lockett, S., Desprez, P. Y. & Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl Acad. Sci. USA 98, 12072–12077 (2001).
Cahu, J., Bustany, S. & Sola, B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 3, e446 (2012).
Canino, C. et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 31, 3148–3163 (2012).
Ohanna, M. et al. Secretome from senescent melanoma engages the STAT3 pathway to favor reprogramming of naive melanoma towards a tumor-initiating cell phenotype. Oncotarget 4, 2212–2224 (2013).
Parrinello, S., Coppe, J. P., Krtolica, A. & Campisi, J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. 118, 485–496 (2005).
Gaston-Massuet, C. et al. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc. Natl Acad. Sci. USA 108, 11482–11487 (2011).
Andoniadou, C. L. et al. Sox2(+) stem/progenitor cells in the adult mouse pituitary support organ homeostasis and have tumor-inducing potential. Cell Stem Cell 13, 433–445 (2013).
Muller, H. L., Merchant, T. E., Puget, S. & Martinez-Barbera, J. P. New outlook on the diagnosis, treatment and follow-up of childhood-onset craniopharyngioma. Nat. Rev. Endocrinol. 13, 299–312 (2017).
Buslei, R. et al. Common mutations of beta-catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol. 109, 589–597 (2005).
Kato, K. et al. Possible linkage between specific histological structures and aberrant reactivation of the Wnt pathway in adamantinomatous craniopharyngioma. J. Pathol. 203, 814–821 (2004).
Sekine, S. et al. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am. J. Pathol. 161, 1997–2001 (2002).
Brastianos, P. K. et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat. Genet. 46, 161–165 (2014).
Hofmann, B. M. et al. Nuclear beta-catenin accumulation as reliable marker for the differentiation between cystic craniopharyngiomas and rathke cleft cysts: a clinico-pathologic approach. Am. J. Surg. Pathol. 30, 1595–1603 (2006).
Holsken, A. et al. Adamantinomatous craniopharyngiomas express tumor stem cell markers in cells with activated Wnt signaling: further evidence for the existence of a tumor stem cell niche? Pituitary https://doi.org/10.1007/s11102-013-0543-8 (2013).
Stache, C. et al. Insights into the infiltrative behavior of adamantinomatous craniopharyngioma in a new xenotransplant mouse model. Brain Pathol. https://doi.org/10.1111/bpa.12148 (2014).
Boult, J. K. R. et al. Preclinical transgenic and patient-derived xenograft models recapitulate the radiological features of human adamantinomatous craniopharyngioma. Brain Pathol. https://doi.org/10.1111/bpa.12525 (2017).
Sharpless, N. E. & Sherr, C. J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 15, 397–408 (2015).
Zhu, J. & You, C. Craniopharyngioma: survivin expression and ultrastructure. Oncol. Lett. 9, 75–80 (2015).
Sharma, A., Singh, K. & Almasan, A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol. Biol. 920, 613–626 (2012).
Bensimon, A., Aebersold, R. & Shiloh, Y. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett. 585, 1625–1639 (2011).
Li, M. & Yu, X. The role of poly(ADP-ribosyl)ation in DNA damage response and cancer chemotherapy. Oncogene 34, 3349–3356 (2015).
Debacq-Chainiaux, F., Erusalimsky, J. D., Campisi, J. & Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 4, 1798–1806 (2009).
Lee, B. Y. et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 5, 187–195 (2006).
Kurz, D. J., Decary, S., Hong, Y. & Erusalimsky, J. D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 113, 3613–3622 (2000).
Traenckner, E. B. et al. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 14, 2876–2883 (1995).
Samulowitz, U. et al. Human endomucin: distribution pattern, expression on high endothelial venules, and decoration with the MECA-79 epitope. Am. J. Pathol. 160, 1669–1681 (2002).
Andoniadou, C. L. et al. Identification of novel pathways involved in the pathogenesis of human adamantinomatous craniopharyngioma. Acta Neuropathol. 124, 259–271 (2012).
Yosef, R. et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 7, 11190 (2016).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016).
Kennedy, A. L. et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol. Cell 42, 36–49 (2011).
Mosteiro, L. et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 354 https://doi.org/10.1126/science.aaf4445 (2016).
Garcia-Lavandeira, M. et al. Craniopharyngiomas express embryonic stem cell markers (SOX2, OCT4, KLF4, and SOX9) as pituitary stem cells but do not coexpress RET/GFRA3 receptors. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/jc.2011-2187 (2011).
Arzt, E., Chesnokova, V., Stalla, G. K. & Melmed, S. Pituitary adenoma growth: a model for cellular senescence and cytokine action. Cell Cycle 8, 677–678 (2009).
Muller, H. L. et al. Longitudinal study on growth and body mass index before and after diagnosis of childhood craniopharyngioma. J. Clin. Endocrinol. Metab. 89, 3298–3305 (2004).
Kostadinov, S., Hanley, C. L., Lertsburapa, T., O’Brien, B. & He, M. Fetal craniopharyngioma: management, postmortem diagnosis, and literature review of an intracranial tumor detected in utero. Pediatr. Dev. Pathol. 17, 409–412 (2014).
Uhrbom, L., Hesselager, G., Nister, M. & Westermark, B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 58, 5275–5279 (1998).
Fomchenko, E. I. et al. Recruited cells can become transformed and overtake PDGF-induced murine gliomas in vivo during tumor progression. PLoS ONE 6, e20605 (2011).
Chen, Y. et al. Glioma initiating cells contribute to malignant transformation of host glial cells during tumor tissue remodeling via PDGF signaling. Cancer Lett. 365, 174–181 (2015).
Nicolas, M. et al. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 33, 416–421 (2003).
Deschene, E. R. et al. beta-Catenin activation regulates tissue growth non-cell autonomously in the hair stem cell niche. Science 343, 1353–1356 (2014).
Arwert, E. N. et al. Tumor formation initiated by nondividing epidermal cells via an inflammatory infiltrate. Proc. Natl Acad. Sci. USA 107, 19903–19908 (2010).
Kode, A. et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 506, 240–244 (2014).
Baker, D. J. et al. Naturally occurringp16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Shibata, H. et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science 278, 120–123 (1997).
Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L. & Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Keane, T. M. et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294 (2011).
Jones, D. et al. cgpCaVEManWrapper: simple execution of CaVEMan in order to detect somatic single nucleotide variants in NGS data. Curr. Protoc. Bioinform. 56, 10.11–15.10.18 (2016).
Acknowledgements
We sincerely apologise for not including relevant publications due to editorial restrictions. We thank the Developmental Studies Hybridoma Bank (University of Iowa) and the National Hormone and Peptide Program (Harbor–University of California, Los Angeles Medical Center) for providing some of the antibodies used in this study. We thank Dr. Alicia Villa-Osaba for technical help with triple immunostaining experiments. This work was supported by the Medical Research Council (MRC) (Grants MR/M000125/1 and MR/L016729/1), Great Ormond Street Hospital for Children Charity/Children with Cancer UK (GOSHCC/CWCUK) (Grant W1055) and by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children National Health Service Foundation Trust and University College London. J.M.G.-M. is supported by a CONACYT-UCL Postgraduate Fellowship and a CONACYT-REDESD travel fellowship. S.H. and S.P. are supported by a Wellcome Trust Ph.D. Fellowship. J.R.A. is supported by a Cancer Research UK Clinical Research Training Fellowship. D.J.A. and M.R. are supported by Cancer Research UK, The Wellcome Trust and the ERC Synergy Programme. J.P.M.-B. is a Great Ormond Street Hospital for Children’s Charity Principal Investigator.
Author information
Authors and Affiliations
Contributions
J.M.G.-M. carried out the majority of the experiments, contributed to experimental design. S.H., G.C., S.P., S.N.M.-G. and C.S. performed experiments. J.A. and N.J. carried out the bioinformatics analysis on RNA sequencing data and contributed to the analysis of the exome sequencing data. G.K. carried out the immunostainings shown in Fig. 3. L.P. genotyped mice. A.G. performed the ablation experiments using the ABT senolytic compounds on IMR90 cells. A.V. and T.S.J. contributed to the pathological analysis of human samples. M.R. and D.J.A. performed the exome sequencing of mouse tumours and the bioinformatics analysis. C.L.A. performed and analysed experiments. J.G., C.R.G. and C.L.A. contributed to the overall experimental design and provided intellectual and strategic input. J.P.M.-B. designed experiments and overviewed the project. J.M.G.-M. and J.P.M.-B. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gonzalez-Meljem, J., Haston, S., Carreno, G. et al. Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun 8, 1819 (2017). https://doi.org/10.1038/s41467-017-01992-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-01992-5
This article is cited by
-
METTL3 promotes cellular senescence of colorectal cancer via modulation of CDKN2B transcription and mRNA stability
Oncogene (2024)
-
Pituitary stem cells: past, present and future perspectives
Nature Reviews Endocrinology (2024)
-
Pituitary tumours without distinct lineage differentiation express stem cell marker SOX2
Pituitary (2024)
-
Aging microenvironment and antitumor immunity for geriatric oncology: the landscape and future implications
Journal of Hematology & Oncology (2023)
-
COPI vesicle formation and N-myristoylation are targetable vulnerabilities of senescent cells
Nature Cell Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
