
Abstract
Hainan Island, located between East Asia and Southeast Asia, represents an ideal region for the study of the genetic architecture of geographically isolated populations. However, the genetic structure and demographic history of the indigenous Tai-Kadai-speaking Hlai people and recent expanded southernmost Han Chinese on this island are poorly characterized due to a lack of genetic data. Thus, we collected and genotyped 36 Qiongzhong Hlai and 48 Haikou Han individuals at 497,637 single nucleotide polymorphisms (SNPs). We applied principal component analysis, ADMIXTURE, symmetrical D-statistics, admixture-f3 statistics, qpWave, and qpAdm analysis to infer the population history. Our results revealed the East Asian populations are characterized by a north-south genetic cline with Hlai at the southernmost end. We have not detected recent gene flow from neighboring populations into Hlai, therefore, we used Hlai as an unadmixed proxy to model the admixture history of mainland Tai-Kadai-speaking populations and southern Han Chinese. The mainland Tai-Kadai-speaking populations are suggested deriving a larger number of their ancestry from Hlai-related lineage, but also having admixture from South Asian-related or other neighboring populations. The Hlai group is also suggested to contribute about half of the ancestry to Han Chinese in Hainan. The complex patterns of genetic structure in East Asia were shaped via language categories, geographical boundaries, and large southward population movements with language dispersal and agriculture propagation.
Similar content being viewed by others

Introduction
The archeological record in East Asia documents the complex historical details of past human population movements and occupations with climate and subsistence strategy changes, the origin and spread of agriculture, and technological and cultural changes [1,2,3,4,5]. The earliest archaic hominin activity occurred here over 1.6 million years ago, and the first anatomically modern humans lived here at least 50,000 years ago [6]. The origin of agriculture in the Yangzi River Basin (rice) and Yellow River Basin (foxtail millet and broomcorn millet) and the subsequent expansion of corresponding farming economies northward into the West Liaohe River Basin and southward into Southeast Asia significantly changed the genetic legacy of southern China and Southeast Asia and the gene pool of the Island Southeast Asia and Oceania with Austronesian expansion [5]. Understanding the genetic legacy and demographic history of ethnolinguistically, geographically and culturally diverse Asian populations is a complex and important task that requires whole-genome sequencing or high-density array chip data to represent and illuminate environmental, cultural and geographic range diversity. Previous whole-genome studies on the Han Chinese population highlighted the significant genetic distinction between Northeast Asia and Southeast Asia [7]. A recent autosomal genetic study based on over 10 thousand samples via low-coverage whole-genome sequencing revealed slight east-west genetic divergence [8]. Knowledge of the genetic diversity and population structure of East Asians has also been significantly improved via the HGDP Project, the Simons Genome Project, the 1000 Genomes Project, the Human Genome Diversity Project and the HapMap Project [9,10,11,12]. However, significant gaps in coverage still exist in the southernmost and smallest Hainan province. Populations residing on Hainan Island have been underrepresented in genome-wide-based studies of human population history reconstruction and genetic diversity exploration, since only 50,000 single nucleotide polymorphisms (SNPs) in 10 Jiamao samples have been studied in the Pan-Asian Project before. The population history of the Tai-Kadai-speaking populations in southern China is still very much in its infancy.
Hainan Island, located between Southeast Asia and East Asia and separated by the Qiongzhou Strait from the Leizhou Peninsula, is the 42nd largest island in the world, with a population size of 9,257,600. The ethnic group composition of the island comprises Han Chinese (84%), Tai-Kadi-speaking Hlai (also referred as Li, 14.7%), Hmong-Mien-speaking Miao (0.7%), and Tai-Kadai-speaking Zhuang (0.6%) peoples. Archeological data from Sanya (Hainan) suggested that anatomically modern humans settled on Hainan Island more than 20,000 years ago, in Paleolithic times [13]. Evidence from the paternal genetic structure based on Y-STRs and/or Y-SNPs in Hainan aborigines suggested that Hainan indigenous Hlai people diverged from Mainland Chinese people before 20 kya [14]. Song et al. recently found that the founding paternal lineage of Hlai groups was O1b1a1a1a1a1b-CTS5854. Allele frequency divergence of this lineage between the Hlai population and surrounding populations further supported the small amount of gene flow among them [2]. Li et al. found a high frequency of the Hainan aborigine-dominant haplogroups O1a* and O2a* and a low frequency of the Mainland East Asian-dominant haplogroup O3, suggesting that these isolated populations underwent little genetic admixture during recent population migration events [14]. However, maternally inherited markers have shown genetic similarity between island and mainland peoples in East Asia [15, 16]. To shed more light on the demographic history of Tai-Kadai-speaking populations and Han Chinese individuals from the southernmost part of China, we conducted comprehensive genome-wide population genetic analysis via principal component analysis (PCA), model-based population structure analysis ADMIXTURE, Fst, and formal tests of admixture in ADMIXTOOLS (f3, D, qpWave and qpAdm) [17, 18] and phylogenetic relationship and split events reconstruction approaches (TreeMix, MDS and N-J tree) [19] by using genome-wide data consisting of ~497,637 SNPs from 84 islanders and three sample sets combining publicly available data for various geographic and linguistic affiliations [11, 12, 18, 20].
Materials and methods
The detailed methods are described in Supplementary Note One. Eighty-four unrelated samples were collected with written informed consent. These samples enrolled in the present study were collected randomly from unrelated participants whose parents and grandparents are indigenous people and have the non-consanguineous marriage of the same ethnical group at least three generations. Affymetrix WeGene V1 arrays were used to genotype 497,637 SNP markers. We combined our data with previously published genome-wide data to form three datasets. PCA was carried out using Plink v1.9 and smartPCA [21, 22]. Model-based clustering analysis was performed via ADMIXTURE [17]. Formal tests (f3, D, qpWave, and qpAdm) were performed using ADMIXTOOLS [18]. Calculation of pairwise Fst genetic distances was performed using Plink v1.9 [22]. Mitochondrial and Y-chromosome haplogroup assignments were made using an in-house script. A neighbor-joining tree was constructed using TreeMix version 1.12 [19] and Mega 7.0 [23]. The variation data reported in this paper have been deposited in the Genome Variation Map (GVM) in Big Data Center [24], Beijing Institute of Genomics (BIG), Chinese Academy of Science, under accession numbers GVM000053 that are publicly accessible at http://bigd.big.ac.cn/gvm/getProjectDetail?project=GVM000053.
Results
Population structure of Tai-Kadai-speaking Hlai and southernmost Han Chinese
We successfully genotyped 497,637 genome-wide SNPs after quality control in 36 Hlai and 48 Han individuals in the southernmost province in China (Fig. S1). We obtained three different datasets by merging our new data with previously published population genome-wide data retrieved from the literature [11, 12, 18, 20, 25]. Dataset 1 harbors 346,634 SNP genetic variations in 2048 individuals of 69 populations mainly from HGDP and HapMap3 projects. Dataset 2 has 15,660 genetic variants in 2012 PanAsian individuals from 76 populations. Dataset 3 has 120,894 SNP genetic variants in 2152 individuals of 169 populations from the Human Origin dataset.
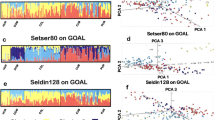
We first carried out the PCA. Variation explained by the top three components in the PCA based on dataset 1 grouped the Hlai and Han individuals with the East Asian cluster (Figs. S2 and 3), which resembled the continental worldwide geographical distribution. The PCA performed only on the 26 East Asian populations showed two genetic affinity clines (south-north cline and east-west cline) and a strong association with language family affiliation (Fig. 1a, b). The south-north cline consisted of Sinitic-, Tibeto-Burman-, Chinese Austroasiatic-, Hmong-Mien- and Tai-Kadai-speaking populations, and the east-west cline included Tungusic-, Mongolic- and Turkic-speaking populations. Our newly studied Hlai population was located at the end of the south-north cline. The Haikou Han individuals were located in an intermediate position between the Hlai and northern Han Chinese (Beijing Han) individuals and had a close genetic relationship with southern Chinese minorities, such as the Jing and Dai groups. Our observed genetic affinity clines were further supported by the PCA results inferred from the pan-Asian dataset and genetic variations in Chinese and adjacent populations (Figs. S4 and 5). Thus, we identified a relatively close genetic relationship between the Tai-Kadai-speaking Hlai and Austronesian-speaking Ami and Atayal groups (Fig. S5).

Genetic affinity inferred from South-North Cline and East-West Cline based on the first two components (a) and the second and third components (b); c Ancestry proportions of Hlai and Han residing in Hainan Island. Plots representing ancestry components of two studied populations and 67 worldwide present-day populations, showing converged runs of 7 (No substructure emerges in East Asia), 8 (Substructure emerges in East Asia) and 13 (Best predefined k value dissected the genetic structure of our sample set).
To subsequently dissect different genetic ancestral components at the individual level in the two newly studied groups and explore the deeper East Asian substructure patterns or potential gene flow, we performed unsupervised model-based ADMIXTURE analyses based on dataset 1 and the pan-Asian dataset (Figs. 1c and S6). We found global populations clustered according to geographical distribution. East Asian populations were grouped in a way that was consistent with linguistic classification and affiliation. The best-fitting model for the worldwide reference populations was based on k = 13 (Fig. S7). We observed two highly specific genetic components among East Asians: one northern ancestry component (saffron yellow, here defined as Northeast Asian: NEA) was mostly represented in Altai-speaking (the Daur, Hezhen, Oroqen, Mongolian and Yakut populations) and Japanese populations and was also detected in the Uyghur and Hazara populations. The proportion of this NEA ancestry decreased gradually from north to south. The other was the southern ancestry component (light green, here defined as Southeast Asian: SEA) highly enriched in the Tai-Kadai-speaking Dai, Hlai, and Austroasiatic-speaking Cambodian and Jing groups. The proportion of this SEA component decreased gradually from south to north. These observed north-south structuring patterns were also apparent in the other predefined ancestry sources (k > 7, Fig. S6) and regional model-based clustering results of subpopulations from dataset 1 (Fig. S7). We found the Qiongzhong Hlai individuals were genetically differentiated from other Asians with minimum NEA ancestry, however, the Haikou Han individuals’ ancestry was characterized by a considerable amount of NEA component (Figs. S8–10).
To further explore the genetic affinities and validate the aforementioned patterns of relatedness between Han, Hlai and other global reference populations, we reanalyzed our genome-wide data together with data from 166 other global populations from seven geographic regions included in the human origin dataset (Dataset 3). PCA revealed that our two investigated populations were located at the end of the Eurasian genetic cline and maintained a close genetic relationship with East Siberian and East Asian populations (Fig. S11). Similar patterns of genetic affinity were obtained after removing the non-East Asian populations. PC1 and PC2, which maximized the genetic differences between Northeast Asians and Southeast Asians, differentiated the Qiongzhong Hlai population as a genetically southern population (Fig. S12). The Hlai population was located between the Ami and Dai populations, while the Haikou Han population was positioned intermediately among the Lahu, Han, Dai, and Kinh populations. Population ancestry dissection via the human origin array data not only revealed significant genetic differentiation between the Hlai and Han populations but also demonstrated a large proportion of shared ancestry between them (Figs. S13 and 14). In summary, the Hlai population was relatively isolated and formed one independent cluster. The Haikou Han Chinese individuals were surrounded by Tai-Kadai, Tibeto-Burman, Hmong-Mien and Austroasiatic speakers in the plot. This observed genetic pattern suggested that southernmost Han Chinese individuals share ancestry with surrounding populations, while the Hlai people are quite homogeneous and probably represent the unadmixed form of Tai-Kadai-speaking populations.
Phylogenetic relationships between two Hainan populations and global reference populations
To characterize the genetic differentiation between the two studied Hainan populations and other reference populations, we first calculated pairwise Fst genetic distances (Table S1) and visualized the results using a heatmap (Fig. S15). The Qiongzhong Hlai population had a close genetic relationship with the Haikou Han population (Fst: 0.0058). However, the Haikou Han population had a close genetic relationship with the Han population (0.0020). We subsequently reconstructed the phylogenetic relationships between the two Hainan populations and global or Asian reference populations using TreeMix or Mega based on the first two datasets. A phylogenetic tree without admixture events strongly supported significant genetic assimilation within Tai-Kadai-speaking populations (the Hlai, Jing, and Dai population). We also found that northern Han Chinese populations were genetically closely related to Northeast Asians, clustering among Japanese, Lahu, other Tibetan-Burman-speaking populations and Altai-speaking populations in the maximum likelihood tree (Fig. S16). When including more populations from Asia based on the reference populations from the Pan-Asian Project, the Qiongzhong Hlai population first clustered with the Jiamao population and then with the Zhuang and four Tai populations, forming the Tai-Kadai-speaking population cluster. The Haikou Han population first clustered with the Chinese Singapore and Minnan populations and then with the Cantonese population, forming the Han Chinese branch (Figs. S17–19). The smallest genetic distances among these groups further supported the observed genetic affinity (Table S2). Consistent patterns of pairwise genetic distances were also observed among the populations based on the human origin dataset (Fig. S20). Based on Fst genetic distances (Table S3), the Haikou Han and Qiongzhong Hlai populations first grouped together and then grouped with the Tai-Kadai-speaking Dai and Austronesian-speaking Ami and Atayal populations (Fig. S21).
We further calculated outgroup-f3-statistics in the form f3(A, B; Yoruba) to explore genetic relationships (Table S4–6). As shown in Fig. 2a, b, the Qiongzhong Hlai population shared an excess of alleles with three Jing populations, the Haikou Han population and the Dai population (f3: 0.2273–0.2280), while the Haikou Han population shared the most genetic drift with the Qiongzhong Hlai population (0.2278), followed by the Han population (0.2262). Heatmap results based on the outgroup-f3 statistic of dataset 1 showed striking genetic affiliation among our two studied groups and the Jing, Dai, and Lahu populations (Figs. S22 and 23). Here, we defined two outgroup f3-based genetic distances: 1-f3 and 1/f3. Multidimensional scaling (MDS) plots based on the two abovementioned genetic distances suggested a significant genetic affinity between the Qiongzhong Hlai and Haikou Han populations (Fig. S24). The phylogenetic relationship reconstruction based on 1-f3 showed a close phylogenetic relationship among the Haikou Han, Hlai, Jing, and Dai populations, which was consistent with the patterns observed based on the heatmap and MDS (Fig. S25). Shared genetic drift based on the pan-Asian dataset showed more shared genetic history between the Ami and Haikou Han populations, as well as among the Qiongzhong Hlai, Ami and Zhuang populations (Figs. S26–28). We also identified a close genetic relationship between the Qiongzhong Hlai population and other Tai-Kadai-speaking Zhuang and Jiamao, adjacent Haikou Han and Minnan Han populations via MDS and a neighbor-joining tree (Figs. S29 and 30). Further supporting evidence for genetic similarity based on our focuses and references are also provided by a variety of statistics derived from the genetic variations obtained with the human origin dataset (Figs. S31–33).

a,b Outgroup f3-statistics (three-population tests) in the form f3 (X, Haikou Han; Yoruba) or (X, Qiongzhong Hlai; Yoruba), where X is a reference population from Dataset1. A higher value (red) denotes higher shared genetic drift between our studied populations and the included present-day reference populations. c–e D-statistics in the form of D(studied populations, Dai/Haikou Han/Beijing Han; X, Yoruba) shows the shared branch length between two investigated populations or Dai/Haikou Han/Beijing Han and all worldwide tested populations. Negative D values with significant Z-scores indicate that the included reference population shares more derived alleles with Dai/Haikou Han/Beijing Han populations than with others, and positive D values with significant Z-scores indicate that the tested population shares more alleles with studied populations than with others. The absolute value of Z-score larger than 3 is considered as statistically significant.
Modeling the Tai-Kadai-speaking population and southern Chinese affinity
To explore the phylogenetic affiliation between the newly investigated populations and Eurasian references, we subsequently calculated D-statistics(Asians, non-Africans; Qiongzhong Hlai, Yoruba) (Supplementary Note Two, Tables S7–13) and found excess genetic affinity between the Qiongzhong Hlai and East Asian-related populations. This pattern was further validated via all possible D-statistics in the form D(Qiongzhong Hlai, East Asians; non-Africans, Yoruba). Figure 2c, d shows the statistics for D(Qiongzhong Hlai, Dai;/Qiongzhong Hlai, Haikou Han; X, Yoruba). We observed positive D values with Z-scores > 3 in 10 East Asian populations for D(Qiongzhong Hlai, Dai; X, Yoruba), showing that the Japanese, Beijing Han, Tokyo Japanese, Denver Chinese, Han, She, three Jing and Haikou Han populations shared more alleles with the Qiongzhong Hlai population than with the Dai population, and other populations had symmetrical genetic relationships with the two populations. In contrast, South Asians (Sindhi and Gujarati Indian) shared more alleles with the Dai population than with the Hlai population (Fig. 2c), suggesting that South Asians contributed some ancestry to the Dai population after the separation of the Hlai and Dai. We next calculated admixture-f3-statistics (source 1, source 2; Dai) to explore the potential source populations. Surprisingly, no negative f3 values were observed (Table S17). Similar results were obtained with the human origin dataset. However, the smallest f3 values were observed between the Qiongzhong Hlai and Sindhi populations (0.484 standard error, SE), followed by Gujarati Indians (0.754 SE), suggesting Dai population may derive some ancestry from South Asian related lineages.
When focusing on geographically close and linguistically similar populations, as shown in Figs. 2d and S34–36, we subsequently found that the Dai, Cambodian and three Jing groups all shared more genetic drift with the Qiongzhong Hlai population than with Sinitic-, Hmong-Mien-, and Tibeto-Burman-speaking populations (the Haikou Han, Denver Chinese, Han, She, Miao and Tujia populations). Only the Haikou Han population shared significantly more alleles with the Qiongzhong Hlai population than with the Austroasiatic Jing population; however, all East Asian, American, Melanesian, Central Hazara and Uyghur groups shared more genetic drift with the Hlai population than with Austroasiatic-speaking Cambodians (Fig. S37), suggesting that Cambodians may have some deep ancestry. We did not find a population sharing significantly more alleles with the Hlai population than with the Austronesian-speaking Atayal and Ami populations (Figs. S38–S41), suggesting that the Hlai and Atayal/Ami populations could form a clean clade without influence from surrounding populations.
We similarly used f3-statistics in the form of f3(source 1, source 2; Qiongzhong Hlai) to find the ancestral populations of the Hlai population (Table S14). We did not detect signals of potential admixture in Hlai individuals (Fig. S42, all f3 values larger 0.0039; Tables S15 and 16), which was consistent with the isolated position of the Hlai population in the PCA and unique ancestry component observed in the ADMIXTURE analysis. Thus, Hlai ancestry could be on Hainan Island in an unadmixed form since this population diverged from the common ancestor of proto-Tai-Kadai speakers.
We further used the qpWave and qpAdm packages in ADMIXTOOLS to test the fit of the mixture model of the Dai population and estimate the proportion of prespecified ancestry sources. QpWave/qpAdm modeling can be used to integrate a series of D/f-statistics and was developed to test whether there are asymmetrical relationships among multiple outgroups, a putative admixture model and focused targets. Here, we used the Mbuti Pygmy, Yoruba, Papuan, Melanesian, Basque, Sardinian and Karitiana groups as the outgroups. We modeled the Dai population as a mixture of two or three divergent ancestry sources: an ancient ancestral Tai-Kadai-related source (here represented by the Qiongzhong Hlai population), ancient ancestral South Asian-related source (represented by Sindhi or Gujarati Indians), and ancient ancestral East Asian mainland-related source (represented by the Tibeto-Burman-speaking Yi, Hmong-Mien-speaking Miao or Sinitic-speaking Han population).
Our qpWave results showed that a two-way admixture model could be used to elucidate the observed genetic variation in the Dai population (p_rank1 > 0.05). The mainland Dai population harbored a large proportion of Qiongzhong Hlai-related ancestry, with smaller ancestry proportions from South Asians (98.6% Hlai + 1.4% Gujarati Indians: p_rank1 = 0.578; 98.9% Hlai + 1.1% Sindhi: p_rank1 = 0.511). We also considered the significant impact of the neighboring ethnic groups on the formation of the Dai gene pool. We subsequently used three-way admixture models to model the detailed population history of the Dai population. The Dai population was modeled as 78.2% derived from Hlai-related, 0.2% from Sindhi-related and 21.6% from Tibeto-Burman-speaking Yi-related ancestries (p_rank2 = 0.587). Using Hmong-Mien-related or Sinitic-related populations as mainland sources, we documented a similar ancestry proportion composition (87.7% derived from the Hlai population, 1.1% from the Sindhi population and 11.2% from the Miao population, p_rank2 = 0.230; 93.9% derived from the Hlai population, 1.1% from the Sindhi population and 5.1% from the Han-related population, p_rank2 = 0.345).
Admixture history of the southernmost Han Chinese population
To formally explore the genomic formation of the southernmost Haikou Han population, we performed a series of three- or four-population tests concentrated on the Haikou Han population. The Z-score of D-statistics in the form D(Beijing Han, Qiongzhong Hlai; Haikou Han, Yoruba) = −10.197 and D(Dai, Qiongzhong Hlai; Haikou Han, Yoruba) = −8.45 indicated that the Haikou Han population was genetically more similar to the Qiongzhong Hlai population than to the Beijing Han or mainland Tai-Kadai-speaking populations (Fig. 2e). Table S18 presents the results for D(Asian populations, global populations; Haikou Han, Yoruba), which demonstrated that the Haikou Han population shared an extra affinity with Tai-Kadai populations or other southern Chinese populations when compared to other populations. We also obtained consistent evidence that the Haikou Han population shared more alleles with southern populations from D(Haikou Han, Asian populations; global populations, Yoruba) when the “Asian population” was represented by Hmong-Mien-, Sinitic-, Tibeto-Burman-, Turkic-, Tungusic- and Mongolic-speaking populations (Figs. S43–48). However, if we compared the Haikou Han population with Austroasiatic or Austronesian-speaking populations in the form of D(Haikou Han, Austroasiatic or Austronesian-speaking populations; X, Yoruba), we found that the Haikou Han population shared some ancestry with northern populations, such as the Beijing Han, Hezhen, Daur, and Yakut populations, among others (Figs. S49 and 50).
To explore the promising ancestral source-related populations of Haikou Han Chinese individuals, we subsequently calculated 2778 sets of admixture-f3-statistics in the form f3(source 1, source 2; Haikou Han) (Table S19). Three population testing resulted in significant negative f3 values with absolute Z-scores larger than 3 when the Qiongzhong Hlai population was regarded as one source and the northern East Asian population (especially for Beijing Han: Z = −30.987 or Hezhen: Z = −26.389) was used as the second potential ancestry source. We also observed statistically significant f3-statistics when northern East Asian Tungusic speakers and southern East Asian Tai-Kadai or Austroasiatic speakers were used as the two targeted source populations. Tables S20 and 21 provide the results of admixture-f3-statistics based on the pan-Asian and human origin datasets, which consistently suggested that the Qiongzhong Hlai population and one northern population served as the best possible pair of sources. The result was consistent with the intermediate position of the Haikou Han population on the north-south genetic cline in the qualitative PCA plots and the shared Hlai-dominant and northern East Asian-dominant ancestries in ADMIXTURE. We then used qpWave to model the minimum number of ancestry sources for the Haikou Han population and used qpAdm to evaluate the corresponding ancestry coefficients. The qpWave results indicated that the Haikou Han population could be described by a two-way admixture model (χ2p = 0.3260), with ancestry from two sources related to the Qiongzhong Hlai (0.552) and Beijing Han (0.448) populations. We also used the two-way admixture model of Southeast Asian ancestry (Qiongzhong Hlai-related: SEAA) and Northeast Asian ancestry (Beijing Han-related: NEAA) to characterize populations with a close relationship to the Haikou Han population (Table S22): Han (χ2p = 0.089; SEAA:NEAA = 0.317:0.683), Shanxin Jing (χ2p = 0.3576; SEAA:NEAA = 0.787:0.213), Wanwei Jing (χ2p = 0.6469; SEAA:NEAA = 0.724:0.276), Wutou Jing (χ2p = 0.3225; SEAA:NEAA = 0.68: 0.32), Miao (χ2p = 0.3786; SEAA:NEAA = 0.377:0.623), She (χ2p = 0.1275; SEAA:NEAA = 0.344:0.656) and Tujia (χ2p = 0.7754; SEAA:NEAA = 0.185: 0.815). Genetic differences between the Hlai and Han populations were also identified via the uniparental genetic landscape and are presented in Supplementary Note Three (Table S23).
Ancient genetic admixture or introgression in East Asia
To further explore the possible genetic introgression between archaic hominins (Denisovan and Neanderthals) and the genetic relationship between East Asian modern and ancient peoples, we merged our data with genomic data from chimpanzees, Altai Denisovan and Mezmaiskaya Neanderthals, 66 ancient and 656 present-day East Eurasian individuals included in the human origin dataset [1, 3, 26,27,28] (Fig. 3a). When the ancient samples were projected onto the aforementioned genetic landscape, the ancient genomes from Southeast Asia were projected near Austronesian, Austroasiatic or Tai-Kadai speakers. Interestingly, Vietnamese individuals from the late Neolithic or Bronze Age clustered closely to the studied Qiongzhong Hlai population. We subsequently used ADMIXTURE to model the two studied populations in combination with the ancient peoples with five predefined ancestral source populations (Fig. 3c). We observed three ancestry components in our studied Hlai and Haikou Han. One ancestral component with maximum representation in the Mlabri people and widely distributed in the mainland southeastern ancient samples, representing Austroasiatic ancestry, was observed in a small proportion in the Qiongzhong Hlai and Haikou Han populations. One ancestral component with maximum representation in the Tungusic-speaking Ulchi population and Devil’s Cave population and representing the Northeast Asian lineage (NEAL) was identified as having a larger proportion in the Haikou Han population and a smaller proportion in the Qiongzhong Hlai population. The remaining ancestry component (yellow) in our two studied populations was maximized in Austronesian speakers and enriched in ancient samples from Southeast Asia, which represented an approximate source of the Southeast Asian lineage (SEAL).

a Geographical locations of included modern and ancient populations or individuals; b Results of two-dimensional plots based on top components, ancient people are projected; c ADMIXTURE result shows the ancestry components of modern and ancient people when five ancestral sources are predefined; d Heatmap showed the pairwise shared alleles estimated via outgroup-f3 (Source1, Source2; Yoruba); e Modeling the ancestral source of Tai-Kadai-speaking Dai, Hlai and Haikou Han Chinese via qpWave and qpAdm.
To formally test the genetic affinity between and find the potential ancestral sources of modern and ancient East Asian peoples, we measured allele sharing and admixture signals via outgroup-f3 and admixture-f3 statistics. The obtained results (Fig. 3d) showed strong genetic affinity among the ancient populations from historic times in Southeast Asia (Vietnam_Historical, Philippines_RedSlippedPottery and Malaysia_Historical), which was consistent with the patterns observed via PCA and ADMIXTURE analysis. The admixture-f3 results (Tables S24~25) further illustrated that one ancient or modern NEAL (Devil’s Cave line) combined with one SEAL (Atayal, Vietnamese Neolithic or Hoabinhian lineages) or an ancient/modern Tibetan-associated lineage combined with the SEAL could provide a better fit of the admixture model for the Haikou Han population, but we have not found well-fitting model for the Qiongzhong Hlai population. We finally used one typical NEAL (DevilsCave_N) combined with the SEAL from different times (a hunter-gatherer lineage represented by the Laos_Hoabinhian or Onge group, which formed one clade, and the Neolithic people from Vietnam) as two sources. The targeted populations could be explained well with two sources of ancestry: the Devil’sGate-related and the Lao_Hoabinhian-related groups, with respective proportions of 73.8 and 26.2% (the Qiongzhong Hlai population), 79.6 and 20.4% (Haikou Han), and 72.3 and 27.7% (Dai); the Devil’s Cave-related and Onge-related groups (79.4 and 20.6% (Hlai), 84.4 and 15.6% (Han), and 77.6 and 22.4% (Dai)); and the Devil’s Cave-related and more recent Vietnam_N-related groups (68.6 and 31.4% (Hlai), 75.6 and 24.4% (Han), and 67.2 and 32.8% (Dai)).
Finally, potential admixture between archaic hominins and Hlai, Han and other East Asian populations was estimated via formal tests of f4 statistics (French, East Asian; Yoruba, Archaic). We have not found significant signals for the Haikou Han and Hlai populations in the above test. Furthermore, Z-score of the f4(Qiongzhong Hlai, Haikou Han; Vindija_light, Primate_Chimp) = 0.747 and Z-score of f4(Qiongzhong Hlai, Haikou Han; Denisova_light, Primate_Chimp) = −1.577 showed a symmetrical relationship of Hlai and Han with regard to archaic ancestry. To further determine the archaic admixture proportion, we calculated the f4-ratio in the form f4(Vindija Neanderthal, Chimpanzee; X, Yoruba)/f4(VindijaNea, Chimpanzee; Mezmaiskaya Neanderthal, Yoruba). Han Chinese individuals inherited 4.12% of their genome from Neanderthals and exhibited low levels of Denisovan ancestry (~0.0317%). Hlai individuals harbored 3.81% Neanderthal ancestry and no Denisovan introgression based on f4(Denisovan, Yoruba; X, Han_Taiwan)/f4(Denisovan, Yoruba; Papuan, Han_Taiwan). Due to the relatively low density of this merged dataset, archaic introgression in Hainan islanders based on high-coverage sequencing data should be assessed in further population genetic studies.
Discussion
Southern China is considered the genetic and geographic origin of Tai-Kadai-speaking populations [29, 30]. There have been subsequent continuous migrations southward from Yunnan and Guangxi provinces into Southeast Asia (with populations mainly settling in Thailand and Vietnam) for ~1000 years since the middle Neolithic expansion period, such as the southward migration of millet farmers from the Ganqing region [1, 3]. In addition, the cultural patterns and settlement history of the middle and lower Yangtze Valley illustrated that southern China (Qujialing-Shijiahe and Liangzhu cultures) is an agricultural origin center. Two waves of southward migration with rice agriculture further enhanced the genetic diversity in southern China and Southeast Asia: coastal dispersal to mainland southeastern China and Taiwan and inland dispersal to southwestern China and Southeast Asia are associated expansions of Austronesian- and Austroasiatic-speaking populations [31], respectively. In this study, we comprehensively analyzed high-density autosomal markers in Qingzhong Hlai and Haikou Han populations in combination with corresponding regional or global publicly available whole-genome data to test hypotheses regarding the genetic and geographic origins and dispersal of Tai-Kadai-speaking populations (Fig. 4). Genetic studies focused on mitochondrial sequences, Y-chromosomal SNPs/STRs and autosomal STRs have recently investigated the genetic relationships and structure of different populations (including Han and Hlai populations) on Hainan Island [2, 16, 32, 33]. A finer-scale population demographic history of geographically isolated populations of Hainan needs to be investigated due to the limited number or the relatively low resolution of markers employed in previous studies. We presented a genome-scale genetic analysis of Haikou Han and Qiongzhong Hlai populations and provided the first high-density SNP evidence for the genetic structure, admixture history, and gene flow of these two southernmost populations. This study provided genetic evidence for the phylogenetic affinity between Austronesian and Tai-Kadai speakers, which tends to support the linguistic hypothesis that Kradai and Austronesian language families belonging to the Austro-Tai linguistic phylum [34].

The model was constructed on the basis of our observed genetic ancestry information and previous findings in archeological, linguistic and genetic findings.
Admixture history of the southernmost Han Chinese population
In agreement with the origin of Sino-Tibetan language families in North China and the southward spread of the Sinitic language group, a lot of archeological and genetic evidence has supported the Han Chinese population originated from Neolithic millet farmers in North China [4, 35]. A recent whole-genome sequence study suggested that Sinitic-speaking Hans and Tibeto-Burman-speaking Tibetans diverged more than 9000 kya [4]. A previous Y-chromosome study also provided genetic evidence for the separation between Hans and Tibetans in paleolithic times and that they experienced population expansion ~6 kya [35]. Modern Han Chinese individuals are widely distributed in East Asia due to advances in economic change and subsistence strategies in Neolithic and historic times. Xu and Chen et al. performed the first two genome-wide genetic studies in 2009 and revealed genetic substructure between northern Han Chinese and southern Han Chinese [36]. Mitochondrial and Y-chromosomal evidence also revealed sex-biased admixture in the southern Han Chinese population from northern Han Chinese and southern minorities [37]. In the present study, we estimated the Haikou Han group derived 55.2% of their ancestry from the Hlai-related ancestral population and 44.8% of their ancestry from the Beijing Han-related ancestral population (Table S22). Although persistent barriers to gene flow between the earliest-arriving southern Han Chinese and southern native minority groups may have existed during the early stages of settlement and subsequent migration, the genetic assimilation observed in this study further suggested that modern southern Han Chinese resulted from extensive genetic admixture between Han Chinese and southern aborigines. This pattern of Han Chinese population history is consistent with the demic diffusion model [37]. We also identified a west-east cline caused by genetic variation from Tibeto-Burman and Turkic-speaking populations (Fig. 4), showing the influence from western Eurasians on Turkic-speaking groups in northern China.
The Hainan Han population is the southernmost Han Chinese population. The Han people migrated to Hainan Island in the past 2000 years since 46 BC (https://en.wikipedia.org/wiki/Hainan). Hainan aborigines probably lived on the island from more than 7000 years ago [13]. The common residence history of two thousand years shaped the current genetic structure of the Hainan Han Chinese population. Autosomal STR-based results suggested that the Hainan Han population had a closer genetic relationship with neighboring Han Chinese populations, such as the Taiwan Han population, than with adjacent minority groups [33]. The phylogenetic relationships reconstructed based on genetic variation in Y-STRs illustrated that the Hainan Han population had a striking genetic affinity with the Tai-Kadai-speaking Zhuang population, which suggests potential genetic admixture with nearby Kadai-speaking populations [32]. Song et al. also found that the O2-M122 haplogroup was the dominant haplogroup in their studied Han population [2]. Our comprehensive population genomic results revealed a genetic affinity between the Haikou Han and Qiongzhong Hlai populations. The Han population was located close to the Hlai population in the PCA plots based on genetic variability in the HGDP, HapMap3, pan-Asian and human origin datasets, which is subsequently supported by the maximum likelihood-based ADMIXTURE, pairwise Fst, and neighbor-joining relationship analyses. QpWave and qpAdm together modeled the modern Hainan Han population as a mixture of two components genetically related to the Qiongzhong Hlai and Beijing Han populations: approximately half of their ancestry was from the Beijing Han-related population, and a half was derived from a Qiongzhong Hlai-associated source. Taken together, these results revealed the fine-scale genetic structure of Han Chinese people residing on Hainan Island. The two-way admixture model of the Haikou Han population proposed in this study suggested that the complex admixture of Han Chinese populations occurred on this island due to the Han Chinese expansion and subsequent admixture with surrounding indigenous peoples.
Genetic structure of Tai-Kadai-speaking populations
The Tai-Kadai languages, also referred to as the Kam-Tai, Kra-Dai, Daic and Kadai languages, harbors ~70 language groups. The Tai-Kadai-speaking populations are widely distributed in southern China, the mainland of Southeast Asia and South Asia, with a population of more than 2 million that stretches from Hainan Island in the east to Northeast India in the west and from southern Sichuan in the north to southern Thailand in the south. Our study provided new insights into the genetic history of Tai-Kadai-speaking populations in China. In sum, the Qiongzhong Hlai population exhibits an unadmixed genetic structure without detectable recent influence from surrounding populations, indicating that it can be used as the proxy for pro-Tai-Kadai speakers. We also observed genetic affinity among the Qiongzhong Hlai and Austronesian-speaking Ami and Atayal populations. The observed genetic connection between Tai-Kadai speakers and Austronesian speakers is consistent with the common origin of these two language families in mainland China during the Neolithic period before rice agriculture expanded toward the south via the coastal route (Fig. 4).
The genetic composition of Hlai was less affected by the Neolithic farming expansion or historical migration compared with other mainland Tai-Kadai populations. We modeled the genetic admixture history of Tai-Kadai-speaking populations residing in mainland China using the Hlai population and other potential ancestral populations. Our results from qpWave and qpAdm analyses indicated that the Tai-Kadai-speaking Dai population is a mixture of Hlai-related ancestral populations (~99%) and South Asian-related populations (1%). The small proportion of South Asian related ancestry in the Dai people may be caused by the Austroasiatic language and agricultural spread between East Asia and South Asia [38]. Geographically close ethnic groups may also play an important role in forming the gene pool of the Dai population, especially the Sinitic-speaking Han, Tibeto-Burman-speaking Yi, and Hmong-Mien-speaking Miao peoples.
Previous ancient genomic studies established the genetic stability and continuity in Northeast Asia and the Tibetan Plateau [27, 28]. The 7600year-old Devil’s Cave people and 2000~3000-year-old Nepalese people provided the best ancient references for reconstructing the population history of East Asians. In addition, ancient peoples that lived in prehistoric and historic times (Hoabinhian, Neolithic, Bronze Age and Iron Age peoples) in Southeast Asia provide another excellent Southeast Asian ancestral reference for studying the past in East Asia [1, 3]. Ancient signals in Hainan Islanders based on northern and southern ancient genetic variations further illustrated that genetic affinity was associated with geography and linguistic boundaries. We also found that ancestral sources from northern lineages (the Devil’s Cave people) played a more important role in shaping the genetic landscape of Southeast Asians, which may have been influenced by the southward expansion of millet farmers via two propagation paths (western and central corridors) and rice farmers via coastal and inland routes [31, 39]. Integrating the genetic information from high-density autosomal and paternally and maternally inherited markers is a precise, powerful and convincing method with which to dissect population structure, elucidate sex-biased migrations, analyze socio-cultural or linguistic effects and reconstruct human population history. Compared with previous studies based on genetic variation in Y-chromosomal STRs and SNPs or forensic autosomal genetic markers of STRs, the current assessment incorporating three components (autosomal markers and two types of uniparentally inherited markers) facilitated a better understanding of the origin, migration events and subsequent genetic admixture of Tai-Kadai-speaking populations, especially the Hainan Hlai population. At the same time, genetic findings from the presented study illustrated a complex pattern of genetic admixture and migration history in Tai-Kadai-speaking populations. Thus, more fine-scale Asian population history can be reconstructed when ancient DNA from Hainan ancient people and neighboring South Chinese paleolithic, Neolithic and Bronze Age populations were successfully extracted and reported.
Conclusions
We concluded that the Han Chinese population dispersed southward onto Hainan Island and admixed with the Tai-Kadai-speaking Hlai population, forming the modern southernmost Han Chinese gene pool with 55.2% ancestry from northern Hans and 44.8% from southern minorities. The Qiongzhong Hlai population shows a unique homogeneous genetic structure without obvious admixture with surrounding or incoming populations, which was probably caused by the long-term isolation that occurred since the population’s separation from ancestral Tai-Kadai-speaking populations. The Hlai people are suggested to be survivors of a homogeneous lineage closely related to the ancestor of rice farmers from the Yangzi River Basin or the ancestor of proto-Tai-Kadai speakers in Southeast Asia, whereas the Tai-Kadai-speaking populations in mainland China or Southeast Asia were largely admixed, with ancestry from other surrounding groups. In addition, both ancient and modern genomes illustrated strong associations between genetic affinity and geographical and linguistic categories. Holocene populations that spread southward (the NEAL, millet and rice farmers) significantly shaped the genetic landscape of southern peoples.
References
Lipson M, Cheronet O, Mallick S, Rohland N, Oxenham M, Pietrusewsky M, et al. Ancient genomes document multiple waves of migration in Southeast Asian prehistory. Science. 2018;361:92–5.
Song M, Wang Z, Zhang Y, Zhao C, Lang M, Xie M, et al. Forensic characteristics and phylogenetic analysis of both Y-STR and Y-SNP in the Li and Han ethnic groups from Hainan Island of China. Forensic Sci Int Genet. 2019;39:e14–e20.
McColl H, Racimo F, Vinner L, Demeter F, Gakuhari T, Moreno-Mayar JV, et al. The prehistoric peopling of Southeast Asia. Science 2018;361:88–92.
Lu D, Lou H, Yuan K, Wang X, Wang Y, Zhang C, et al. Ancestral origins and genetic history of Tibetan highlanders. Am J Hum Genet. 2016;99:580–94.
Chen FH, Dong GH, Zhang DJ, Liu XY, Jia X, An CB, et al. Agriculture facilitated permanent human occupation of the Tibetan Plateau after 3600 B.P. Science. 2015;347:248–50.
Higham C. Hunter-gatherers in southeast Asia: from prehistory to the present. Hum Biol. 2013;85:21–43.
Liu S, Huang S, Chen F, Zhao L, Yuan Y, Francis SS, et al. Genomic analyses from non-invasive prenatal testing reveal genetic associations, patterns of viral infections, and Chinese population history. Cell. 2018;175:347–59 e14.
Chiang CWK, Mangul S, Robles C, Sankararaman S. A comprehensive map of genetic variation in the World’s largest ethnic group-Han Chinese. Mol Biol Evol. 2018;35:2736–50.
Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature. 2016;538:201–6.
Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81.
Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, Ramachandran S, et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–4.
Consortium, HP-AS, Abdulla MA, Ahmed I, Assawamakin A, Bhak J, Brahmachari SK, et al. Mapping human genetic diversity in Asia. Science 2009;326:1541–5.
Li Y, Hao S, Huang W, Forestier H, Zhou Y. Li H. Luobi Cave, South China: A Comparative Perspective on a Novel Cobble-Tool Industry Associated with Bone Tool Technology during the Pleistocene–Holocene Transition. J World Prehistory. 2019;32:143–78.
Li D, Li H, Ou C, Lu Y, Sun Y, Yang B, et al. Paternal genetic structure of Hainan aborigines isolated at the entrance to East Asia. PLoS One 2008;3:e2168.
Li DN, Wang CC, Lu Y, Qin ZD, Yang K, Lin XJ, et al. Three phases for the early peopling of Hainan Island viewed from mitochondrial DNA. J Syst Evol. 2013;51:671–80.
Peng MS, He JD, Liu HX, Zhang YP. Tracing the legacy of the early Hainan Islanders-a perspective from mitochondrial DNA. BMC Evol Biol. 2011;11:46.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–64.
Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, et al. Ancient admixture in human history. Genetics. 2012;192:1065–93.
Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8:e1002967.
The International HapMap C, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007;449:851.
Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet 2006;2:e190.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Song S, Tian D, Li C, Tang B, Dong L, Xiao J, et al. Genome variation map: a data repository of genome variations in BIG data center. Nucleic Acids Res. 2017;46(D1):D944–D9.
Huang X, Zhou Q, Bin X, Lai S, Lin C, Hu R, et al. The genetic assimilation in language borrowing inferred from Jing People. Am J Phys Anthropol. 2018;166:638–48.
Yang MA, Gao X, Theunert C, Tong H, Aximu-Petri A, Nickel B, et al. 40,000-year-old individual from Asia provides insight into early population structure in Eurasia. Curr Biol. 2017;27:3202–8 e9.
Jeong C, Ozga AT, Witonsky DB, Malmstrom H, Edlund H, Hofman CA, et al. Long-term genetic stability and a high-altitude East Asian origin for the peoples of the high valleys of the Himalayan arc. Proc Natl Acad Sci USA. 2016;113:7485–90.
Sikora M, Pitulko VV, Sousa VC, Allentoft ME, Vinner L, Rasmussen S, et al. The population history of northeastern Siberia since the Pleistocene. Nature. 2019;570:182–8.
Kutanan W, Kampuansai J, Srikummool M, Brunelli A, Ghirotto S, Arias L, et al. Contrasting paternal and maternal genetic histories of Thai and Lao populations. Mol Biol Evol. 2019;36:1490–506.
He G, Wang Z, Zou X, Wang M, Liu J, Wang S, et al. Tai-Kadai-speaking Gelao population: forensic features, genetic diversity and population structure. Forensic Sci Int Genet. 2019;40:e231–e9.
Chi Z. Hung H-c. The emergence of agriculture in southern China. Antiquity. 2010;84:11–25.
Wang M, Wang Z, Zhang Y, He G, Liu J, Hou Y. Forensic characteristics and phylogenetic analysis of two Han populations from the southern coastal regions of China using 27 Y-STR loci. Forensic Sci Int Genet. 2017;31:e17–e23.
Wang M, Wang Z, He G, Jia Z, Liu J, Hou Y. Genetic characteristics and phylogenetic analysis of three Chinese ethnic groups using the Huaxia Platinum System. Sci Rep. 2018;8:2429.
Blench R, Sagart L, Sanchez-Mazas A. The peopling of East Asia: putting together archaeology, linguistics and genetics. 2005; 136–60.
Wang LX, Lu Y, Zhang C, Wei LH, Yan S, Huang YZ, et al. Reconstruction of Y-chromosome phylogeny reveals two neolithic expansions of Tibeto-Burman populations. Mol Genet Genom. 2018;293:1293–300.
Xu S, Yin X, Li S, Jin W, Lou H, Yang L, et al. Genomic dissection of population substructure of Han Chinese and its implication in association studies. Am J Hum Genet. 2009;85:762–74.
Wen B, Li H, Lu D, Song X, Zhang F, He Y, et al. Genetic evidence supports demic diffusion of Han culture. Nature. 2004;431:302–5.
Tatte K, Pagani L, Pathak AK, Koks S, Ho Duy B, Ho XD, et al. The genetic legacy of continental scale admixture in Indian Austroasiatic speakers. Sci Rep. 2019;9:3818.
He K, Lu H, Zhang J, Wang C, Huan X. Prehistoric evolution of the dualistic structure mixed rice and millet farming in China. Holocene. 2017;27:1885–98.
Acknowledgements
CCW was supported by the National Natural Science Foundation of China (31801040), Nanqiang Outstanding Young Talents Program of Xiamen University (X2123302), and Fundamental Research Funds for the Central Universities (ZK1144). YH was supported by grants from the Opening Project of Shanghai Key Laboratory of Crime Scene Evidence (2016XCWZK12), the National Natural Science Foundation of China (81571854) and the Fundamental Research Funds for the Central Universities (20826041A4408, YJ201651, and 2012017yjsy187).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
He, G., Wang, Z., Guo, J. et al. Inferring the population history of Tai-Kadai-speaking people and southernmost Han Chinese on Hainan Island by genome-wide array genotyping. Eur J Hum Genet 28, 1111–1123 (2020). https://doi.org/10.1038/s41431-020-0599-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0599-7
This article is cited by
-
Differentiated adaptative genetic architecture and language-related demographical history in South China inferred from 619 genomes from 56 populations
BMC Biology (2024)
-
Extensive genetic admixture between Tai-Kadai-speaking people and their neighbours in the northeastern region of the Yungui Plateau inferred from genome-wide variations
BMC Genomics (2023)
-
Multiple founding paternal lineages inferred from the newly-developed 639-plex Y-SNP panel suggested the complex admixture and migration history of Chinese people
Human Genomics (2023)
-
Phylogenetic evidence reveals early Kra-Dai divergence and dispersal in the late Holocene
Nature Communications (2023)
-
The genomic history of southwestern Chinese populations demonstrated massive population migration and admixture among proto-Hmong–Mien speakers and incoming migrants
Molecular Genetics and Genomics (2022)
