
Abstract
Familial exudative vitreoretinopathy (FEVR) is an inherited retinal disorder, which is primarily characterized by abnormal development of retinal vasculature. In this study, we reported a subject presenting the clinical features of FEVR as well as microcephaly. Screening of the KIF11 gene in this patient revealed a novel heterozygous protein-truncating variant (c.2717del, p.(L906*), NM_004523.3). Segregation analysis in the unaffected parents using Sanger sequencing suggested the variant to be present in a mosaic state in the unaffected mother. KIF11 exon 19 which harbors the variant was amplified from the proband and her father, as well as three different tissues of the mother, followed by amplicon-based deep sequencing. This analysis revealed that the variant is present in different tissues of the mother at various rates, i.e. in blood (16.9%), saliva (20.7%), or skin biopsy-derived fibroblast cells (6.6%). These data demonstrate the importance of deep sequencing in unaffected parents upon detection of a genetic defect in isolated cases to detect possible mosaicisms, enabling a more reliable recurrence risk assessment and thereby improve genetic counseling.
Similar content being viewed by others


Introduction
Familial exudative vitreoretinopathy (FEVR) is a clinically and genetically heterogeneous disorder, primarily characterized by abnormal development of the retinal vasculature [1, 2]. A serious manifestation of FEVR is a falciform retinal fold, a condition leading to visual impairment at young age [3].
Variants in KIF11 have been reported in individuals with FEVR plus microcephaly, as well as in progressive retinal dystrophy and syndromic patients exhibiting microcephaly, lymphedema, chorioretinal dysplasia, and/or intellectual disability [4,5,6,7,8]. KIF11 variants can occur de novo, or be inherited from one of the parents [4,5,6]. Although missense variants have been reported, the majority of KIF11 variants that lead to FEVR are protein-truncating variants [4,5,6, 9]. KIF11 encodes the protein Eg5, which is involved in mitosis [10]. In vitro inhibition of Eg5 resulted in impaired endothelial cell proliferation and migration [11]. Furthermore, kif11 zebrafish mutants showed defects in glial development [12]. These data underscore the importance of Eg5 in angiogenesis and central nervous system development, which may explain the abnormal retinal vasculature development and microcephaly observed in patients carrying variants in KIF11.
In this study, a novel heterozygous protein-truncating variant in KIF11 was identified in a subject presenting FEVR and microcephaly. The variant was detected in mosaic state in the mother. The level of mosaicism in different tissues of the mother was accurately determined by deep sequencing.
Methods
Subjects and clinical evaluation
A subject with FEVR and microcephaly as well as both her unaffected parents participated in this study. The proband underwent ophthalmological and neurological examination. Clinical examinations were followed-up for 17 years. Following the identification of a genetic mosaicism, the mother was also extensively examined with different ophthalmological tests, as well as a measure of head circumference. Written informed consent was obtained from all participants. This study was approved by the local Ethics Committee and conducted according to the tenets of the Declaration of Helsinki.
Genetic analysis
All exons and intron–exon boundaries of the KIF11 gene were screened for potentially causative DNA variants in the proband by Sanger sequencing and a causative variant was identified. Subsequently, exon 19 of KIF11 was amplified from the proband, her father, and three different tissues of her mother. The amplicons were subjected to Sanger sequencing as well as ion semiconductor sequencing (Ion Personal Genome Machine, Thermo Fisher Scientific, Waltham, Massachussetts, USA) as described previously [13]. The genetic analysis are explained in detail in the Supplementary Information. The variant that was detected has been submitted to the LOVD database (https://databases.lovd.nl/shared/genes/KIF11, individual IDs: 00154935 and 00154936).
Results
The proband is a female patient who was born after a normal gestation and delivery. She was found to have nystagmus, convergent strabismus, and mild microcephaly at an early age. At 3 years of age, low visual acuity and strabismus of the right eye was noted. Occlusion of the left eye was tried for a short period without success and strabismus was corrected by surgery. At the age of 7, best corrected visual acuity (BCVA) of the right eye was finger counting and of the left eye 20/50. Neurological examination at the same age disclosed slight psychomotor retardation. A computer tomography (CT) scan showed no structural abnormalities of the brain and ventricles, nor any cerebral calcifications; the skull circumference was 48 cm (<p2; second percentile). Laboratory tests for metabolic disorders and toxoplasmosis were negative. Fundus examination at the age of 9 showed a prominent falciform retinal fold running from the optic disk to the inferior-temporal periphery in the right eye (Fig. 1a). Some retinal vessels were observed outside the fold and no abnormalities were noted in the peripheral retina. Besides those shown in the fundus picture, some white tissue attached to the pars plana was also observed. In the left eye, areas of retinal pigmented epithelium atrophy (Fig. 1b) and an abrupt termination of the temporal retinal vessels was observed in the equatorial area: the more peripheral part of the retina is avascular and shows some local areas of RPE atrophy.

Fundus photographs of the posterior retina of the proband demonstrating FEVR. a Fundus picture of the right eye, showing a prominent falciform retinal fold (indicated with a dashed box) with several retinal vessels (indicated with white arrowheads) extending from the optic disk in inferior-temporal direction to the fundus periphery. The posterior retina is included in the fold and no macula could be detected. The retina outside the fold is attached and shows some retinal vessels. b Fundus picture of the left eye reveals areas of retinal pigmented epithelium (RPE) atrophy (indicated with a solid box) around the optic disk and along the inferior-temporal vessels. The macula (indicated with a white arrowhead) is slightly ectopic
During 17 years of follow-up, no significant changes in the anterior segments and fundi of both eyes were noted, nor did visual acuity decline. The patient died at 28 years of age by a cause that is unrelated to the phenotype described here.
Since the clinical features present in the proband resemble those of FEVR and microcephaly, KIF11 was Sanger sequenced from blood DNA to detect potentially causative DNA variants. A novel heterozygous one base-pair deletion that immediately results in premature termination of the protein (c.2717del; p.(L906*), hg19, NM_004523.3) was detected (Fig. 2a–c). While the variant was not present in the father (Fig. 2c), small peaks of the mutant allele were detected in the mother’s blood DNA, suggesting a mosaicism. To further investigate this finding, KIF11 exon 19 which harbors the variant was amplified from blood-derived DNA from the proband and her father, as well as DNA of three different tissues from the mother (peripheral blood lymphocytes, skin biopsy-derived fibroblasts, and saliva), followed by amplicon-based deep sequencing using the Ion Personal Genome Machine. For each sample, more than 200,000 reads were obtained. This analysis confirmed that the variant was present heterozygously (45.5% of the reads) in the proband (Fig. 2c) and absent in the father. It also revealed that the variant was present in the mother’s blood, saliva, and fibroblasts at a frequency of 16.9%, 20.7%, and 6.6%, respectively. This not only confirms that the mother is mosaic for this variant, but also demonstrates that the degree of mosaicism can differ between tissues (Fig. 2c).

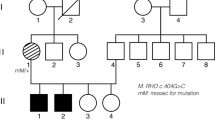
Genetic analysis of KIF11 in a patient with FEVR and microcephaly. a Pedigree of the family described in this study. The affected proband is depicted with a filled symbol. Slash indicates deceased person. M denotes germ line variant, m denotes mosaic variant. b Schematic representation of the KIF11 gene and the encoded Eg5 protein (adapted from Ostergaard et al. [7]). The variant identified in this study is depicted above the scheme. Other KIF11 variants reported in subjects with FEVR and microcephaly cases are indicated below the protein [4,5,6, 9]. cDNA positions corresponding to the indicated protein variants are listed in Table 1. c Sequencing results of the KIF11 variant. Sanger sequencing electropherograms are shown in the upper panel, whereas the relative amount of reads obtained from ion semiconductor sequencing are depicted below in bar graphs. The percentage of reads per given nucleotide is defined as the amount of reads called for a base among the total reads that cover that specific nucleotide. The percentage of reads having the c.2717del variant are indicated above the graph
Following the identification of the mosaicism in the mother, a detailed clinical re-examination was performed, at an age of 57. At a young age, no visual or ocular problems had been reported. When she was 18 years old, she started wearing contact lenses to correct a light myopia of both eyes. Operations for cataract with lens implantation were done in both eyes at age 53. Ophthalmological examination showed normal best corrected visual acuity (right eye: 20/30 and left eye: 20/25). Her head circumference was 55 cm and thereby falls within the normal range. The eye position was straight and good binocular functions were present. Both anterior segments showed a well-positioned artificial lens with minimal opacification of the posterior lens capsule. Slitlamp-biomicroscopy showed posterior poles with a normal configuration of the macula and the vascular arcades of the retina. The peripheral retina was completely vascularized and did not show any dystrophic signs. In conclusion, no signs of FEVR or microcephaly were present in the mother of the proband.
Discussion
A heterozygous variant in KIF11 (c.2717del, p.(L906*)) was detected in a patient presenting FEVR and microcephaly. It was inherited from the mother, who carries the variant in mosaic state at various rates in different tissues (blood, fibroblasts, and saliva). Besides the identification of the genetic defect in this patient, this study also demonstrates the importance of genetic testing in parents and provides insight in the required amount of Eg5, the protein encoded by KIF11, for normal development.
To date, two suspected KIF11 mosaic cases have been reported in literature [4, 5]. Robitaille et al. reported two siblings carrying identical KIF11 variants that were not detected in the healthy parents by Sanger sequencing [4], whereas Hu et al. reported a case in which a mosaicism was suspected via subcloning experiments [5]. To our knowledge, this is the first report in which a KIF11 mosaicism is accurately detected in different tissues of an individual using amplicon-based deep sequencing. This technique may allow the detection of mosaicism that occurs at a low rate which may be undetectable by Sanger sequencing. The presence of the variant in germ cells and somatic cells of the mother indicates that the variant has arisen early in embryonic development, prior to primordial germ cell differentiation (before ~15 mitotic divisions) [14]. Although it cannot be assessed experimentally, we can assume a similar degree of mosaicism present in the germline compared to those detected in saliva and blood, which contain cells originating from ectoderm and mesoderm, respectively. Thereby, we can also hypothesize on the chance of offspring being affected. Thus, the robust detection of mosaicism in unaffected parents can allow more accurate recurrence risks assessments and thereby facilitate better genetic counseling.
In this study, the mosaicism was quantified in three different tissues populated by cells originating from mesoderm (blood and fibroblasts) and ectoderm (saliva). Leukocytes (mesoderm) contamination in the saliva sample may occur [15]. The presence of the variant in these tissues corresponds with the abnormal development of retinal vasculature and microcephaly observed in the patient, which are defects in the development of organs predominantly populated by cells originating from mesoderm and ectoderm, respectively.
A difference in the mosaicism rate was detected in blood vs. fibroblasts, despite both originating from mesoderm. Although this may indicate that the variant occurred after further mesoderm specification, its presence in the germ cells of the mother suggests that the difference is more likely due to an artifact of the in vitro culturing of fibroblasts, which has been shown previously in lymphoblastoid cell lines [15]. Therefore, fresh materials which have not undergone any in vitro culturing should be used to detect mosaicisms.
KIF11 variants reported in subjects with FEVR and microcephaly are equally distributed throughout the Eg5 protein (Fig. 2b) [4,5,6, 9], further highlighting the importance of both the N-terminal and C-terminal part of Eg5 for proper functioning in spindle localization, as shown previously [10]. The variant that is identified in this study lies in the bimC box, a sequence motif surrounding a potential phosphorylation site at the C-terminus of the protein. It is known that Eg5 phosphorylation at this site (specifically at threonine residue 927) is required for its mitotic spindle association during mitosis [10, 16]. The data obtained in this study also allow the speculation of the amount of Eg5 required for a cell to function normally. Assuming a full loss-of-function of the mutated allele and considering the average mosaicism rate detected in the mother (20%), it is tempting to assume that a similar degree of mosaicism is present in other tissues, and thus that ~80% of normal Eg5 protein is sufficient for normal development. Given that a heterozygous variant results in developmental defects, the minimum required amount of Eg5 likely lies between 50% and 80%.
In summary, the findings in this study demonstrate the importance of deep sequencing of parental DNA following the identification of genetic defects in isolated cases to detect possible mosaicisms. It will allow a reliable recurrence risk assessment and thereby improve genetic counseling. It is not only relevant for subjects with FEVR and microcephaly, but also for other genetic disorders with a similar mode of inheritance.
References
Criswick VG, Schepens CL. Familial exudative vitreoretinopathy. Am J Ophthalmol. 1969;68:578–94.
Gilmour DF. Familial exudative vitreoretinopathy and related retinopathies. Eye (London). 2015;29:1–14.
van Nouhuys CE. Congenital retinal fold as a sign of dominant exudative vitreoretinopathy. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1981;217:55–67.
Robitaille JM, Gillett RM, LeBlanc MA, et al. Phenotypic overlap between familial exudative vitreoretinopathy and microcephaly, lymphedema, and chorioretinal dysplasia caused by KIF11 mutations. JAMA Ophthalmol. 2014;132:1393–9.
Hu H, Xiao X, Li S, Jia X, Guo X, Zhang Q. KIF11 mutations are a common cause of autosomal dominant familial exudative vitreoretinopathy. Br J Ophthalmol. 2016;100:278–83.
Li JK, Fei P, Li Y, et al. Identification of novel KIF11 mutations in patients with familial exudative vitreoretinopathy and a phenotypic analysis. Sci Rep. 2016;6:26564.
Ostergaard P, Simpson MA, Mendola A, et al. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am J Hum Genet. 2012;90:356–62.
Birtel J, Gliem M, Mangold E, et al. Novel insights into the phenotypical spectrum of KIF11-associated retinopathy, including a new form of retinal ciliopathy. Invest Ophthalmol Vis Sci. 2017;58:3950–9.
Rao FQ, Cai XB, Cheng FF, et al. Mutations in LRP5, FZD4, TSPAN12, NDP, ZNF408, or KIF11 genes account for 38.7% of Chinese patients with familial exudative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2017;58:2623–9.
Sawin KE, Mitchison TJ. Mutations in the kinesin-like protein Eg5 disrupting localization to the mitotic spindle. Proc Natl Acad Sci USA. 1995;92:4289–93.
Exertier P, Javerzat S, Wang B, et al. Impaired angiogenesis and tumor development by inhibition of the mitotic kinesin Eg5. Oncotarget. 2013;4:2302–16.
Barresi MJ, Burton S, Dipietrantonio K, Amsterdam A, Hopkins N, Karlstrom RO. Essential genes for astroglial development and axon pathfinding during zebrafish embryogenesis. Dev Dyn. 2010;239:2603–18.
Diekstra A, Bosgoed E, Rikken A, et al. Translating sanger-based routine DNA diagnostics into generic massive parallel ion semiconductor sequencing. Clin Chem. 2015;61:154–62.
Campbell IM, Shaw CA, Stankiewicz P, Lupski JR. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet. 2015;31:382–92.
Gajecka M. Unrevealed mosaicism in the next-generation sequencing era. Mol Genet Genom. 2016;291:513–30.
Blangy A, Lane HA, d’Herin P, Harper M, Kress M, Nigg EA. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–69.
Acknowledgements
We gratefully acknowledge the subjects that participated in this study. We would like to thank Dr. Kornelia Neveling and Ronny Derks from the Genomics Technology Center of Radboudumc for assistance with ion semiconductor sequencing, Michael Kwint for assistance with analysis of the ion semiconductor sequencing data, Dr. Alexander Hoischen for helpful discussions on mosaicisms, Dr. Wendy van Zelst-Stams for skin biopsy taking, and Saskia van der Velde-Visser for cell culture assistance.
Funding
This study was financially supported by a Radboudumc PhD Grant to Dyah W. Karjosukarso.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest to report.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Karjosukarso, D.W., Cremers, F.P.M., van Nouhuys, C.E. et al. Detection and quantification of a KIF11 mosaicism in a subject presenting familial exudative vitreoretinopathy with microcephaly. Eur J Hum Genet 26, 1819–1823 (2018). https://doi.org/10.1038/s41431-018-0243-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0243-y
This article is cited by
-
Kinesin-5 Eg5 is essential for spindle assembly, chromosome stability and organogenesis in development
Cell Death Discovery (2022)
