
Abstract
Calreticulin (CALR) is an endoplasmic reticulum (ER)-resident protein involved in a spectrum of cellular processes. In healthy cells, CALR operates as a chaperone and Ca2+ buffer to assist correct protein folding within the ER. Besides favoring the maintenance of cellular proteostasis, these cell-intrinsic CALR functions support Ca2+-dependent processes, such as adhesion and integrin signaling, and ensure normal antigen presentation on MHC Class I molecules. Moreover, cancer cells succumbing to immunogenic cell death (ICD) expose CALR on their surface, which promotes the uptake of cell corpses by professional phagocytes and ultimately supports the initiation of anticancer immunity. Thus, loss-of-function CALR mutations promote oncogenesis not only as they impair cellular homeostasis in healthy cells, but also as they compromise natural and therapy-driven immunosurveillance. However, the prognostic impact of total or membrane-exposed CALR levels appears to vary considerably with cancer type. For instance, while genetic CALR defects promote pre-neoplastic myeloproliferation, patients with myeloproliferative neoplasms bearing CALR mutations often experience improved overall survival as compared to patients bearing wild-type CALR. Here, we discuss the context-dependent impact of CALR on malignant transformation, tumor progression and response to cancer therapy.
Similar content being viewed by others


Introduction
According to current models, oncogenesis involves two main, highly-interconnected components: (1) the accumulation of genetic and/or epigenetic defects that endow (initially) normal cells with an increased proliferative potential, an accrued resistance to cell death, the ability to drive neo-angiogenesis, as well as the capacity to disseminate to form metastases,1 and (2) the escape from local and systemic control by the host immune system (immunoevasion).2,3 Thus, malignant transformation (i.e., the process through which a healthy cell becomes a neoplastic cell precursor) and tumor progression (i.e., the proliferation of such precursor coupled with the acquisition of increasingly malignant features), as well as the response of established tumors to therapy, occur in the context of a bidirectional interaction between (pre-)malignant cells and their host that ultimately dictates clinical outcome.4,5,6,7 In this context, alterations that simultaneously endow cancer cells with an intrinsic proliferative advantage and limit the ability of the immune system to recognize and eliminate them (Box 1) are expected to be potent drivers of oncogenesis.
Calreticulin (CALR) is a Ca2+-binding endoplasmic reticulum (ER) protein that aids the folding of proteins destined to secretion and insertion in the plasma membrane, de facto providing a major contribution to the maintenance of cellular homeostasis when unfolded proteins accumulate within the ER (e.g., in the context of viral infection).8,9,10 Moreover, CALR mechanistically contributes to the initiation of adaptive anticancer immunity in the context of immunogenic cell death (ICD), a functional variant of regulated cell death (RCD) that is sufficient to elicit an antigen-specific immune response in immunocompetent, syngeneic hosts (provided that dying cells express antigens not covered by central or peripheral tolerance).11,12,13 In particular, CALR exposed on the surface of cancer cells undergoing ICD mediates robust pro-phagocytic effects, hence facilitating the uptake of dying cells or their corpses by antigen-presenting cells (APCs), in particular immature dendritic cells (DCs), that migrate to lymph nodes to cross-prime tumor-specific naïve CD8+ T cells.11,12
Thus, wild-type CALR expressed at physiological levels operates at the interface between the preservation of cellular homeostasis and the initiation of an immune response that eradicates cells experiencing damage beyond recovery in support of organismal fitness.14,15 In line with this notion, CALR is mutated or downregulated in a variety of neoplasms.4,16,17 Here, we discuss the cell-intrinsic and immunological mechanisms whereby CALR influences malignant transformation, tumor progression and response to therapy.
CALR in cellular homeostasis
CALR is a highly conserved Ca2+-binding chaperone of 417 amino acids (MW: 46 kDa) that is predominantly localized in the ER lumen.18 CALR consists of three distinct domains: (1) an N-terminal lectin-like globular domain that is responsible for the interaction of CALR with α integrins and contains a steroid receptor-like DNA-binding site involved in chaperone functions; (2) a central proline-rich (P) domain with high-affinity and low-capacity for Ca2+ binding that participates in CALR chaperone activity; and (3) a highly acidic C-terminal region with low-affinity and high-capacity for Ca2+ binding that is involved in CALR Ca2+-buffering functions, followed by a C-terminal KDEL domain that ensures CALR retrieval from the Golgi apparatus to the ER18,19 (Fig. 1a).

a Wild-type calreticulin (CALRWT) mediates key functions not only as it cooperates with CANX and PDIA3 in the control of protein folding within the ER, but also as it contributes to reticular Ca2+ buffering. b Cancer-associated CALR mutations compromise the capacity of mutant CALR (CALRMUT) to be retained in the ER, resulting in anterograde CALR transport from the ER to the plasma membrane via the Golgi apparatus, constitutive association with MPL, and TPO-independent oncogenic signaling (at least in some cells) via JAK2 and MAPKs. P, phosphate.
As a reticular protein, CALR is involved in a quality control system for newly synthesized proteins and glycoproteins that relies on multiple additional chaperones, including (but not limited to) calnexin (CANX), heat shock protein family A (Hsp70) member 5 (HSPA5, also known as BiP or GRP78), heat shock protein 90 beta family member 1 (HSP90B1, also known as GRP94), protein disulfide isomerase family A member 3 (PDIA3).20 Globally, such a system (which is commonly known as the CALR/CANX cycle) prevents the premature export of misfolded proteins from the ER as it supports refolding20 (Fig. 1a), hence occupying a central position in the homeostatic response to the accumulation of unfolded polypeptides.9 Alongside, the Ca2+-binding functions of CALR are central for physiological Ca2+ and integrin signaling in both excitable and non-excitable cells.21,22,23 In particular, a cytosolic pool of CALR is required for integrin-dependent cell adhesion, reflecting the ability of CALR to reversibly bind the KxGFFFKR domain in the cytoplasmic tails of α integrins.23,24 In line with the key role of CALR in whole-body physiology, Calr−/− mice die in utero at day 12.5–16.5 as they fail to absorb the umbilical hernia and manifest prominent cardiac alterations.25 Interestingly, such developmental alterations seem to result from transcriptional defects caused by Ca2+-related nuclear pore complex malfunction,26 rather than from reduced contractility. Consistent with this interpretation, the adult heart expresses low CALR levels25 and transgene-enforced CALR overexpression in the adult heart causes contractility issues that culminate with complete heart block and sudden death.27 Moreover, the Calr−/− genotype imposes functional defects to a variety of cell types beyond cardiomyocytes,28 such as T cells,29 endothelial cells,30 vascular smooth muscle cells,31 fibroblasts,32 and oocytes.33
Recurrent somatic mutations in CALR affecting the majority of patients with myeloproliferative neoplasms (MPNs) who do not bear mutations in Janus kinase 2 (JAK2) and MPL proto-oncogene, thrombopoietin receptor (MPL) were first documented in 2013.34,35 Such CALR mutations most often consist of insertions and/or deletions in exon 9, resulting in a C-terminal domain that bears a novel, positively-charged amino acid sequence and lacks the KDEL domain (Fig. 1b), thus enabling mutant CALR to escape the ER and form stable complexes with MPL by interacting with glycans on MPL N117 residue.36,37 These complexes are exposed on the surface of hematopoietic precursors through normal anterograde ER-to-Golgi transport, culminating with thrombopoietin (TPO)-independent MPL dimerization, consequent JAK2 and mitogen-activated protein kinase (MAPK) activation, and final signal transducer and activator of transcription (STAT) signaling.36,38,39,40 Ultimately, this cascade drives the deregulated clonal expansion of hematopoietic stem cells and megakaryocytes that underlies MPNs, as documented in a variety of animal models.41,42,43 In support of this hierarchical signal transduction model, the oncogenic potential of human CALR mutations is highly compromised upon the depletion of either MPL or JAK2, as well as in the presence of pharmacological JAK2 inhibitors.44,45
Preclinical data suggest that type I CALR mutations, which eliminate all negatively-charged amino acids from the CALR C-terminus (e.g., del52), appear to be superior to their type II counterparts, which only eliminate approximately half of such residues (e.g., ins5), at driving thrombocytosis progression to myelofibrosis,40,46 an oncogenic function that strictly depends on the CALR N-terminal domain.47 Consistent with this notion, patients with type I CALR mutations experience a more aggressive disease course with rapid progression to acute lymphoid leukemia (AML) as compared to individuals with type II mutations.48 Of note, both type I and type II CALR mutations not only promote constitutive MPL signaling to support TPO-independent proliferation, but also compromise (at least to some degree) cellular responses to unfolded protein accumulation and oxidative stress,49 thus promoting oncogenesis via accrued generation of reactive oxygen species (ROS) and consequent genomic instability.50 Moreover, both type I and type II CALR mutations have recently been shown to cause TPO-independent Ca2+ fluxes in megakaryocytes,51 thus promoting uncontrolled proliferation.52 As type I mutations are expected to impair the Ca2+-binding functions of CALR more than their type II counterparts, this latter mechanism may contribute (at least partially) to the differential disease course of patients affected by type I versus type II CALR mutations.
Taken together, these observations delineate a precise molecular pathway whereby genetic defects in CALR support malignant transformation in the hematopoietic system via trophic, cell-intrinsic mechanisms. The loss of wild-type CALR functions, however, is also expected to favor oncogenesis by compromising immunosurveillance, as discussed below.
CALR in antigen presentation
CALR is an integral part of the so-called peptide-loading complex (PLC), a transient multicomponent complex that assembles at the membrane of the ER to ensure the proper loading of cellular antigens onto MHC Class I molecules.53,54,55 Besides CALR, the PLC involves PDIA3, TAP-binding protein (TAPBP, also known as tapasin), transporter 1, ATP-binding cassette subfamily B member (TAP1) and transporter 2, ATP-binding cassette subfamily B member (TAP2), which collectively mediate: (1) the assembly of MHC Class I heavy chains with beta 2 microglobulin (B2M); (2) the ATP-dependent transportation of cytosolic peptides to the ER lumen; (3) the loading of such peptides on the antigen-binding pocket of MHC Class I molecules, and (4) the release of loaded MHC Class I for anterograde ER-to-Golgi transport and exposure on the plasma membrane.56 CANX also participates in peptide loading by binding to (and hence stabilizing) MHC Class I heavy chains prior to their stabilizing interactions with B2M56 (Fig. 2).

CALR, CANX and PDIA3 are key to the proper assembly of MHC Class I heavy chains with B2M in the ER lumen, as well as to (1) the loading of antigenic peptides onto mature MHC Class I molecules and (2) the retrieval of sub-optimally assembled MHC Class I molecules from post-ER compartments, notably the ERGIC and the cis-Golgi. TCR, T-cell receptor.
Specifically, CALR contributes to PLC functions by at least two different mechanisms.57 First, by interacting with PDIA3 in a glycan-dependent manner, CALR preserves steady-state levels of TAPBP and MHC Class I heavy chains.58,59 Second, CALR can retrieve suboptimally assembled MHC Class I molecules from post-ER compartments, notably the ER-Golgi intermediate compartment (ERGIC) and the cis-Golgi.60 Thus, both mouse and human cells lacking CALR exhibit a major (50%–80%) reduction in peptide loading onto MHC Class I molecules and exposure of properly loaded MHC on the cells surface,61 a defect that cannot be rescued by re-expression of CALR variants lacking the C-terminus.62 Consistent with this, cancer-related CALR mutants are unable to support the activity of the PLC and hence are associated not only with constitutive MPL signaling (see above), but also with reduced antigen presentation on MHC Class I molecules,63 de facto favoring immunoevasion upon loss of tumor antigenicity.64 Further supporting this notion, even in the absence of oncogenic mutations, CALR levels are reduced in multiple solid tumors at advanced stage, generally correlating with reduced MHC Class I exposure on the cell surface and poor disease outcome.65,66,67
Apparently at odds with the above, at least some cancer-relevant mutations of CALR exon 9 have been shown to generate shared tumor neoantigens (TNAs) eliciting spontaneous immune responses in patients with MPNs.68,69 Such responses, however, largely rely on antigen presentation by MHC Class II molecules and appear to involve a population of CD4+ T cells with cytolytic functions,68 explaining why they can arise in the context of impaired MHC Class I presentation. Of note, spontaneous CALR-targeting immune responses in MPN patients appear to be under strict regulation by cytotoxic T lymphocyte-associated protein 4 (CTLA4) and programmed cell death 1 (PDCD1, best known as PD-1) signaling.70 Consistent with this, administration of the PD-1-targeting immune checkpoint blocker (ICB) pembrolizumab restored T cell immunity against mutant CALR in some patients with MPN.70 Notably, CD8+ T cell responses were also detected in this setting,70 potentially reflecting the ability of interferon gamma (IFNγ) produced by type I helper (TH1) CD4+ cells to overcome the defect in MHC Class I presentation associated with CALR mutations.71
Taken together, these findings suggest that shared mutations in CALR exon 9 are amenable to targeting by immunotherapy, and a phase I clinical trial testing a therapeutic peptide-based vaccine specific for mutant CALR in subjects with MPN is ongoing (NCT03566446).72 Since CALR mutations are the second most common MPN drivers and the TNAs they generate are shared among different patients, this therapeutic strategy, if successful, may be beneficial for a significant number of patients with MPN.35,73
CALR in danger signaling
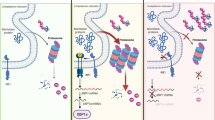
Preclinical and clinical data accumulating over the past decade indicate that besides playing a key role in the maintenance of reticular homeostasis and in antigen presentation, CALR is a major determinant of cellular adjuvanticity, i.e., the ability of stressed and dying cells to deliver co-stimulatory (rather than co-inhibitory) signals to immune cells11,12 (Fig. 3). This function does not originate from physiological CALR pools within the ER or ERGIC, but from an expanded pool of CALR molecules that are exposed on the membrane of cells undergoing the so-called integrated stress response (ISR),74 a multipronged, cell-wide reaction to specific perturbations of the extracellular or intracellular microenvironment that cause (in a majority of cases) the loss of ER homeostasis.75,76 In particular, the translocation of CALR on the outer surface of the plasma membrane, which relies on the inactivating phosphorylation of eukaryotic translation initiation factor 2 subunit alpha (EIF2S1, best known as eIF2α)77 (Box 2), can be induced by a number of chemotherapeutic agents (i.e., anthracyclines, taxanes, oxaliplatin, bortezomib, PT-112, and others),78,79,80,81,82,83,84 radiation therapy,85,86 some variants of photodynamic therapy,87,88,89,90,91 high hydrostatic pressure,92,93 oncolytic peptides,94,95,96 and oncolytic viruses,97,98,99,100,101 among others.102,103

The exposure of CALR on the surface of stressed and dying tumor cells mediate multipronged immunostimulatory effect. First, surface-exposed CALR promotes the uptake of dying cells or their corpses by DCs, a process that is actively inhibited by CD47. Second, the exposure of CALR has been associated with the activation of an IFN response in dying cells, although the underlying mechanisms remain to be elucidated. Third, surface-exposed CALR promotes the expansion of CD11b+CD14+ monocytes that proficiently trans-present IL15 to NK cells. CXCL10, C-X-C motif chemokine ligand 10; IL2RB, interleukin 2 receptor subunit beta; IL2RG, interleukin 2 receptor subunit gamma; PDT, photodynamic therapy; SIRP1A, signal regulatory protein alpha.
Cell surface-exposed CALR delivers potent pro-phagocytic signals to APCs including DCs and their precursors,104,105 de facto initiating the uptake of dying cells or their corpses in the context of immunostimulation.106,107 This process is a conditio sine qua non for the initiation of a tumor-targeting immune response by cancer cells undergoing ICD.11,12 In line with this notion, wild-type mouse cancer cells exposed to an ICD inducer in vitro can be used as vaccines to establish long-term prophylactic immunity against living cancer cells of the same type (in immunocompetent syngeneic mice), but lose their vaccination potential when CALR is lost, downregulated or blocked.74,89,108 Supporting a specific role for surface-exposed (as opposed to reticular) CALR in this setting, surface adsorption of recombinant CALR generally restores the prophylactic power of CALR-depleted cancer cells undergoing ICD in vitro.74,108 Importantly, CALR exposure on the surface of cells undergoing ICD is generally required, but not sufficient, for cell death to be perceived as immunogenic, as dying cells must also express antigens that are not covered by central or peripheral tolerance in a specific host and emit several other adjuvant-like signals, collectively known as damage-associated molecular patterns (DAMPs),109 as they die.11 These signals, which are decoded by pattern recognition receptors expressed by immune cells,110 include, but are not limited to: (1) ATP secretion, favoring the recruitment of DCs and their precursors to sites of ICD, and their activation;111 (2) the release of the nuclear protein high mobility group Box 1 (HMGB1), which mediates immunostimulatory functions;112 (3) the secretion of annexin A1 (ANXA1), which directs the interaction of DCs to dying cells or their corpses;113 and (4) the synthesis and secretion of type I interferon (IFN), which amplifies local immunostimulation via cancer cell-intrinsic and -extrinsic mechanisms.114,115,116,117 Finally, the site of cell death must be compatible with the initiation of an immune response, and hence be scarcely infiltrated by immunosuppressive cells but accessible to myeloid and lymphoid immune effectors.11
The pro-phagocytic effects of surface-exposed CALR have largely been attributed to LDL receptor-related protein 1 (LRP1, best known as CD91) on the surface of APCs.118 However, additional proteins that bind extracellular CALR have been described, including thrombospondin 1 (THBS1), which has been proposed to cooperate with CALR in the regulation of integrin-independent cell adhesion;119,120 complement C1q A chain (C1QA), which appears to harness CALR as a receptor for cell surface binding;121 as well as members of the mannose-binding lectin (MBL) family.122 Of note, both C1QA and lectin, mannose binding 1 (LMAN1, also known as MBL1) have also been proposed to support the CALR-dependent phagocytosis of apoptotic bodies.123,124 The potential involvement of CD91 in these latter processes, however, remains to be clarified.
The pro-phagocytic effects of surface-exposed CALR are counteracted by at least two different mechanisms. On the one hand, cells undergoing caspase-dependent apoptosis generally expose abundant amounts of phosphatidylserine (PS), a phospholipid normally confined to the inner leaflet of the plasma membrane, on their surface.125,126,127 Surface-exposed PS can be rapidly recognized by the receptor jumonji domain-containing 6, arginine demethylase and lysine hydroxylase (JMJD6, best known as PSR), which initiates the rapid, immunologically silent clearance of cell corpses by macrophages.128,129,130 Thus, CALR must be exposed prior to PS for cell death to be perceived as immunogenic.77,131 On the other hand, the phagocytosis of dying cells is actively inhibited by CD47, an integrin-associated protein that operates by binding signal regulatory protein alpha (SIRPA) on the surface of APCs.132,133,134 Thus, the relative levels of surface-exposed CALR and CD47 dictate the ultimate outcome of the physical interaction between dying cells and phagocytes.135 Multiple companies are developing CD47- or SIRPA-targeting antibodies to promote the phagocytosis of dying cancer cells in the context of immunostimulatory signals that support therapeutically relevant tumor-targeting immunity.136 The clinical potential of these agents, however, remains to be clarified.
Interestingly, surface-exposed CALR seems to mediate immunostimulatory effects that are not directly related to the phagocytosis of dying cells. In particular, high levels of CALR on the surface of AML blasts, which correlates with signs of an ongoing ISR, favors the accumulation of a population of CD11b+CD14+ myeloid cells that express high levels of maturation markers and interleukin 15 receptor, alpha chain (IL15RA).137 This endows CD11b+CD14+ myeloid cells with the ability to trans-present interleukin 15 (IL15) to natural killer (NK) cells, de facto boosting their effector functions against AML cells.137,138 Moreover, CALR exposure on the surface of AML cells has been associated with signatures of type I IFN signaling,137,139 and the immunological control of AML cells engineered to constitutively translocate CALR to the outer leaflet of the plasma membrane is abrogated in mice lacking interferon alpha and beta receptor subunit 1 (IFNAR1).139 Although the molecular mechanisms linking CALR exposure and type I IFN signaling in AML blasts have not yet been elucidated, these findings point to a functional link between two different ICD-relevant DAMPs.
Importantly, both type I and type II mutations in exon 9 compromise the KDEL domain of CALR, which normally enables its traffic from the ER to the Golgi apparatus and back with limited surface exposure or extracellular secretion.48 In line with this notion, cancer cells bearing type I CALR mutations (i.e., del52) secrete increased amount of CALR as compared to their wild-type counterparts.140 Moreover, patients with MPNs driven by CALR exon 9 mutations display elevated plasma levels of CALR as compared to healthy individuals.140 Mechanistically, soluble CALR acts as a decoy for CALR receptors in APCs, de facto limiting the uptake of dying cancer cells and their ability to initiate protective immunity in immunocompetent hosts, correlating with the accumulation of immunosuppressive cells in the spleen and peripheral blood.140 CALR secretion as a consequence of KDEL loss also compromises the therapeutic response of AML to immune checkpoint blockers targeting PD-1, at least in mice.140 In summary, while wild-type CALR operates as a key DAMP in the context of ICD, its mutant counterpart prevents the initiation of tumor-targeting immunity, which identifies yet another immunological advantage conveyed by CALR mutations to cancer cells.
Prognostic and predictive value of CALR
Accumulating data lend further support to the notion that wild-type CALR mediates oncosuppressive effects by limiting malignant features and/or enabling the immunosurveillance of developing tumors (Table 1).17
Low CALR levels have been associated with accrued malignant features including hyperproliferation in preclinical models of prostate cancers141 as well as advanced disease stage in 128 patients with urothelial carcinoma.65 Along similar lines, robust CALR expression in diagnostic biopsies have been linked with improved disease outcome in 68 patients with colorectal carcinoma,142 68 subjects with neuroblastoma,143 three independent cohorts of 270, 125 and 23 individuals with non-small cell lung carcinoma (NSCLC),108,144 105 patients with AML,145 30 subjects with osteosarcoma,146 9 patients with glioblastoma,147 as well as in three independent cohorts of 152, 202 and 302 women with ovarian carcinoma.108,148 In many of these settings, high CALR levels correlated with activation of the ISR,145,148 and/or one or multiple signs of ongoing anticancer immunity, including infiltration by CD45RO+ memory T cells in colorectal carcinoma,142 and abundant intratumoral levels of DCs and TH1 CD4+ T cells in NSCLC.144 Similarly, abundant CALR exposure on the surface of malignant cells has been linked to superior disease outcome in two independent cohort of 50 and 20 patients with AML, correlating with increased levels of T cells specific for tumor-associated antigens149 or limited CD47 expression.150 Importantly, these latter studies were performed in chemotherapy-naïve patients,149,150 suggesting that AML blasts can spontaneously expose CALR on their surface (at least in some settings), potentially as a cellular response to oncogenic stress or adverse microenvironmental conditions in the leukemic marrow.151
Apparently at odds with the above, high CALR levels in diagnostic biopsies have also been associated with negative prognostic value in some patient cohorts (Table 1). In particular, robust CALR expression has been correlated with rapid tumor progression and poor disease outcome in 79 patients with gastric carcinoma,152 58 subjects with NSCLC,153 two independent cohorts of 228 and 33 women with breast carcinoma,154,155 two independent sets of 478 and 251 individuals with neuroblastoma,135 two independent cohorts of 80 and 68 patients with pancreatic cancer,156,157 two independent sets of 165 and 30 subjects with bladder carcinoma,135 and two independent cohorts of 92 and 71 patients with mantle cell lymphoma.135 These findings suggest that the intracellular functions of CALR as a key regulator of Ca2+ homeostasis and integrin-dependent signaling may be required for some tumors to progress. Alternatively, the negative prognostic impact of robust CALR expression in some oncological settings may originate from the compensatory overexpression of CD47, as documented in AML, acute lymphocytic leukemia (ALL) and chronic lymphocytic leukemia (CLL) samples.135 Supporting this possibility, elevated CD47 levels have been linked to poor disease outcome in 132 patients with karyotypically normal AML,158 265 women with ovarian carcinoma,159 and 102 individuals with esophageal squamous cell carcinoma.160
Recently, somatic frameshift mutations in CALR exon 9 have been identified in a large proportion of patients with primary myelofibrosis (PMF), essential thrombocythemia (ET) or polycythemia vera (PV) bearing wild-type JAK2 and MPL.35 These patients are younger and exhibit lower hemoglobin levels, decreased leukocytosis and higher platelet counts as compared to their JAK2- and MPL-mutant counterparts.161 Similarly, PMF and ET patients bearing CALR mutations are younger, and display lower incidence of anemia and leukocytosis, lower Dynamic International Prognostic Scoring System (DIPSS) score and reduced frequency of spliceosome mutations, as well as higher platelet counts than patients with wild-type JAK2, MPL and CALR, de facto experiencing improved disease outcome (median survival: 16 vs 2.3 years).162,163 Altogether, these findings document that CALR mutations constitute a positive prognostic factor in patients with MPN and a valuable predictive biomarker for allocating these patients to allogenic hematopoietic stem cell transplantation.164 Consistent with this, CALR mutational status has recently been incorporated into numerous PMF scoring systems, including the MYelofibrosis Secondary to PC and ET Prognostic Model (MYSEC-PM), the Mutation-enhanced International Prognostic Scoring System for transplant-eligible patients (MIPSS70), and the Genetically Inspired Prognostic Scoring System (GIPSS).165,166,167 Conversely, the CALR, JAK2 and MPL mutational status does not appear to influence the survival of patients with ET.161 The immunobiological bases for this discrepancy remain to be elucidated.
CALR mutations yielding a secreted protein that serves as a decoy to saturate CALR receptors, hence subverting the immune recognition of CALR-exposing stressed and dying tumor cells, have also been documented in ~1%–2% of solid neoplasms.140 Although this percentage is insufficient for prognostic or predictive assessments, it is tempting to speculate that such mutations, which preferentially occur in the C-terminus of the protein,140 may subvert immunosurveillance and favor tumor progression.
In summary, CALR appears to influence malignant transformation, disease progression and response to therapy in various tumor types, standing out as a prominent prognostic factor in multiple oncological settings. To harness the prognostic and potentially predictive potential of CALR, however, it will be important to identify and exclude potential confounders, such as CD47 expression.
Concluding remarks
CALR influences a variety of processes that are key to organismal homeostasis, including (but presumably not limited to) protein folding, Ca2+ homeostasis, cellular adhesion, motility, antigen presentation and danger signaling. It is therefore not surprising that loss-of-function CALR mutations favor (at least in some cases) oncogenesis, tumor progression and resistance to treatment.
At least in part, this scenario is reminiscent of tumor protein p53 (TP53).168 TP53 (best known as p53) mediates indeed a variety of oncosuppressive functions that span from the regulation of metabolism, redox homeostasis and cell fate in (pre)neoplastic cells to the initiation of innate and adaptive anticancer immune responses.168 However, p53 is often inactivated as a consequence of point mutations or hyperactivation of the p53-degrading enzyme MDM2 proto-oncogene (MDM2), which (at least theoretically) can be targeted pharmacologically.169,170 Conversely, CALR mutations are small indels that compromise the C-terminus of the protein, and hence appear difficult to target with pharmacological interventions. Potentially, patients with MPNs bearing CALR mutations may benefit from agents that specifically disrupt CALR–MPL interactions.171 However, the development of such molecules is still in its infancy.
Finally, while the cancer cell-intrinsic functions of CALR may support tumor progression at least in some settings, pharmacological inhibitors affecting the reticular functions of CALR employed as systemic agents may have considerable side effects, in line with the key role of the wild-type protein in several adult tissues (see above). This situation is reminiscent of autophagy. Autophagy is an evolutionarily conserved mechanism for the preservation of cellular and organismal homeostasis that some tumors harness in support of disease progression and resistance to treatment.76 However, autophagy is key to the physiological functions of many tissues, notably the brain, which complicates considerably the use of autophagy inhibitors delivered systemically for cancer therapy.172
Thus, for the time being, CALR stands out mostly as a promising prognostic and/or predictive factor (rather than as a therapeutic target), especially for patients with MPNs. As a potential exception, a phase I clinical trial is currently investigating the safety and preliminary efficacy of a therapeutic peptide-based vaccine specific for mutant CALR in subjects with MPN (NCT03566446). Until now, however, therapeutic peptide-based vaccination has demonstrated limited efficacy in clinical settings,173 which dampens (at least to some degree) enthusiasm on the possibility to treat MPN with a peptide-based vaccine specific for mutant CALR. Additional work is urgently required to devise a therapeutic strategy to target CALR in cancer.
References
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Schreiber, R. D., Old, L. J. & Smyth, M. J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011).
Vitale, I. et al. Mutational and antigenic landscape in tumor progression and cancer immunotherapy. Trends Cell Biol. 29, 396–416 (2019).
Fridman, W. H., Zitvogel, L., Sautes-Fridman, C. & Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 14, 717–734 (2017).
Galon, J., Angell, H. K., Bedognetti, D. & Marincola, F. M. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity 39, 11–26 (2013).
Galluzzi, L., Buque, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 28, 690–714 (2015).
Strasser, K. et al. Immunological differences between colorectal cancer and normal mucosa uncover a prognostically relevant immune cell profile. Oncoimmunology 8, e1537693 (2019).
Saito, Y., Ihara, Y., Leach, M. R., Cohen-Doyle, M. F. & Williams, D. B. Calreticulin functions in vitro as a molecular chaperone for both glycosylated and non-glycosylated proteins. EMBO J. 18, 6718–6729 (1999).
Hetz, C. & Papa, F. R. The unfolded protein response and cell fate control. Mol. Cell 69, 169–181 (2018).
Salvagno, C. & Cubillos-Ruiz, J. R. The impact of endoplasmic reticulum stress responses in dendritic cell immunobiology. Int. Rev. Cell Mol. Biol. 349, 153–176 (2019).
Galluzzi, L. et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 8, e000337 (2020).
Galluzzi, L., Buque, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017).
Ren, L. et al. Identification of neoantigen-specific T cells and their targets: implications for immunotherapy of head and neck squamous cell carcinoma. Oncoimmunology 8, e1568813 (2019).
Schcolnik-Cabrera, A. et al. Calreticulin in phagocytosis and cancer: opposite roles in immune response outcomes. Apoptosis 24, 245–255 (2019).
Galluzzi, L., Yamazaki, T. & Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 19, 731–745 (2018).
Merlinsky, T. R., Levine, R. L. & Pronier, E. Unfolding the role of calreticulin in myeloproliferative neoplasm pathogenesis. Clin. Cancer Res. 25, 2956–2962 (2019).
Fucikova, J. et al. Prognostic and predictive value of DAMPs and DAMP-associated processes in cancer. Front. Immunol. 6, 402 (2015).
Michalak, M., Groenendyk, J., Szabo, E., Gold, L. I. & Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 417, 651–666 (2009).
Nakamura, K. et al. Functional specialization of calreticulin domains. J. Cell Biol. 154, 961–972 (2001).
Rutkevich, L. A. & Williams, D. B. Participation of lectin chaperones and thiol oxidoreductases in protein folding within the endoplasmic reticulum. Curr. Opin. Cell Biol. 23, 157–166 (2011).
Milner, R. E. et al. Calreticulin, and not calsequestrin, is the major calcium binding protein of smooth muscle sarcoplasmic reticulum and liver endoplasmic reticulum. J. Biol. Chem. 266, 7155–7165 (1991).
Burns, K., Helgason, C. D., Bleackley, R. C. & Michalak, M. Calreticulin in T-lymphocytes. Identification of calreticulin in T-lymphocytes and demonstration that activation of T cells correlates with increased levels of calreticulin mRNA and protein. J. Biol. Chem. 267, 19039–19042 (1992).
Coppolino, M. G. et al. Calreticulin is essential for integrin-mediated calcium signalling and cell adhesion. Nature 386, 843–847 (1997).
Coppolino, M., Leung-Hagesteijn, C., Dedhar, S. & Wilkins, J. Inducible interaction of integrin alpha 2 beta 1 with calreticulin. Dependence on the activation state of the integrin. J. Biol. Chem. 270, 23132–23138 (1995).
Mesaeli, N. et al. Calreticulin is essential for cardiac development. J. Cell Biol. 144, 857–868 (1999).
Faustino, R. S. et al. Calreticulin secures calcium-dependent nuclear pore competency required for cardiogenesis. J. Mol. Cell Cardiol. 92, 63–74 (2016).
Nakamura, K. et al. Complete heart block and sudden death in mice overexpressing calreticulin. J. Clin. Invest. 107, 1245–1253 (2001).
Jalali, S., Aghasi, M., Yeganeh, B. & Mesaeli, N. Calreticulin regulates insulin receptor expression and its downstream PI3 Kinase/Akt signalling pathway. Biochim. Biophys. Acta 1783, 2344–2351 (2008).
Porcellini, S. et al. Regulation of peripheral T cell activation by calreticulin. J. Exp. Med. 203, 461–471 (2006).
Biwer, L. A. et al. Endothelial calreticulin deletion impairs endothelial function in aged mice. Am. J. Physiol. Heart Circ. Physiol. 318, H1041–H1048 (2020).
Zimmerman, K. A. et al. Calreticulin regulates neointima formation and collagen deposition following carotid artery ligation. J. Vasc. Res. 52, 306–320 (2015).
Van Duyn Graham, L., Sweetwyne, M. T., Pallero, M. A. & Murphy-Ullrich, J. E. Intracellular calreticulin regulates multiple steps in fibrillar collagen expression, trafficking, and processing into the extracellular matrix. J. Biol. Chem. 285, 7067–7078 (2010).
Tokuhiro, K. et al. Calreticulin is required for development of the cumulus oocyte complex and female fertility. Sci. Rep. 5, 14254 (2015).
Nangalia, J. et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 369, 2391–2405 (2013).
Klampfl, T. et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 369, 2379–2390 (2013).
Chachoua, I. et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 127, 1325–1335 (2016).
Pecquet, C. et al. Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic-defective pathogenic TpoR mutants. Blood 133, 2669–2681 (2019).
Kollmann, K. et al. A novel signalling screen demonstrates that CALR mutations activate essential MAPK signalling and facilitate megakaryocyte differentiation. Leukemia 31, 934–944 (2017).
Han, L. et al. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J. Hematol. Oncol. 9, 45 (2016).
Balligand, T. et al. Pathologic activation of thrombopoietin receptor and JAK2-STAT5 pathway by frameshift mutants of mouse calreticulin. Leukemia 30, 1775–1778 (2016).
Elf, S. et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov. 6, 368–381 (2016).
Shide, K. et al. Calreticulin mutant mice develop essential thrombocythemia that is ameliorated by the JAK inhibitor ruxolitinib. Leukemia 31, 1136–1144 (2017).
Lim, K. H. et al. Expression of CALR mutants causes mpl-dependent thrombocytosis in zebrafish. Blood Cancer J. 6, e481 (2016).
Araki, M. et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 127, 1307–1316 (2016).
Nivarthi, H. et al. Thrombopoietin receptor is required for the oncogenic function of CALR mutants. Leukemia 30, 1759–1763 (2016).
Marty, C. et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood 127, 1317–1324 (2016).
Elf, S. et al. Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood 131, 782–786 (2018).
Pietra, D. et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 30, 431–438 (2016).
Salati, S. et al. Calreticulin Ins5 and Del52 mutations impair unfolded protein and oxidative stress responses in K562 cells expressing CALR mutants. Sci. Rep. 9, 10558 (2019).
Vitale, I., Manic, G., De Maria, R., Kroemer, G. & Galluzzi, L. DNA damage in stem cells. Mol. Cell 66, 306–319 (2017).
Di Buduo, C. A. et al. Defective interaction of mutant calreticulin and SOCE in megakaryocytes from patients with myeloproliferative neoplasms. Blood 135, 133–144 (2020).
Marchi, S., Giorgi, C., Galluzzi, L. & Pinton, P. Ca2+ fluxes and cancer. Mol. Cell 78, 1055–1069 (2020).
Blees, A. et al. Structure of the human MHC-I peptide-loading complex. Nature 551, 525–528 (2017).
Sadasivan, B., Lehner, P. J., Ortmann, B., Spies, T. & Cresswell, P. Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity 5, 103–114 (1996).
Kotsias, F., Cebrian, I. & Alloatti, A. Antigen processing and presentation. Int. Rev. Cell Mol. Biol. 348, 69–121 (2019).
Vyas, J. M., Van der Veen, A. G. & Ploegh, H. L. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 8, 607–618 (2008).
Raghavan, M., Wijeyesakere, S. J., Peters, L. R. & Del Cid, N. Calreticulin in the immune system: ins and outs. Trends Immunol. 34, 13–21 (2013).
Wearsch, P. A., Peaper, D. R. & Cresswell, P. Essential glycan-dependent interactions optimize MHC class I peptide loading. Proc. Natl. Acad. Sci. USA 108, 4950–4955 (2011).
Del Cid, N. et al. Modes of calreticulin recruitment to the major histocompatibility complex class I assembly pathway. J. Biol. Chem. 285, 4520–4535 (2010).
Howe, C. et al. Calreticulin-dependent recycling in the early secretory pathway mediates optimal peptide loading of MHC class I molecules. EMBO J. 28, 3730–3744 (2009).
Gao, B. et al. Assembly and antigen-presenting function of MHC class I molecules in cells lacking the ER chaperone calreticulin. Immunity 16, 99–109 (2002).
Liu, C., Fu, H., Flutter, B., Powis, S. J. & Gao, B. Suppression of MHC class I surface expression by calreticulin’s P-domain in a calreticulin deficient cell line. Biochim. Biophys. Acta 1803, 544–552 (2010).
Arshad, N. & Cresswell, P. Tumor-associated calreticulin variants functionally compromise the peptide loading complex and impair its recruitment of MHC-I. J. Biol. Chem. 293, 9555–9569 (2018).
Galluzzi, L., Chan, T. A., Kroemer, G., Wolchok, J. D. & Lopez-Soto, A. The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 10, eaat7807 (2018).
Cathro, H. P. et al. Relationship between HLA class I antigen processing machinery component expression and the clinicopathologic characteristics of bladder carcinomas. Cancer Immunol. Immunother. 59, 465–472 (2010).
Dierssen, J. W. et al. HNPCC versus sporadic microsatellite-unstable colon cancers follow different routes toward loss of HLA class I expression. BMC Cancer 7, 33 (2007).
Noblejas-Lopez, M. D. M. et al. Expression of MHC class I, HLA-A and HLA-B identifies immune-activated breast tumors with favorable outcome. Oncoimmunology 8, e1629780 (2019).
Holmstrom, M. O. et al. The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia 32, 429–437 (2018).
Holmstrom, M. O., Riley, C. H., Svane, I. M., Hasselbalch, H. C. & Andersen, M. H. The CALR exon 9 mutations are shared neoantigens in patients with CALR mutant chronic myeloproliferative neoplasms. Leukemia 30, 2413–2416 (2016).
Cimen Bozkus, C. et al. Immune checkpoint blockade enhances shared neoantigen-induced T-cell immunity directed against mutated calreticulin in myeloproliferative neoplasms. Cancer Discov. 9, 1192–1207 (2019).
Wallach, D., Fellous, M. & Revel, M. Preferential effect of gamma interferon on the synthesis of HLA antigens and their mRNAs in human cells. Nature 299, 833–836 (1982).
Bezu, L. et al. Trial watch: peptide-based vaccines in anticancer therapy. Oncoimmunology 7, e1511506 (2018).
How, J., Hobbs, G. S. & Mullally, A. Mutant calreticulin in myeloproliferative neoplasms. Blood 134, 2242–2248 (2019).
Obeid, M. et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61 (2007).
Pakos-Zebrucka, K. et al. The integrated stress response. EMBO Rep. 17, 1374–1395 (2016).
Rybstein, M. D., Bravo-San Pedro, J. M., Kroemer, G. & Galluzzi, L. The autophagic network and cancer. Nat. Cell Biol. 20, 243–251 (2018).
Panaretakis, T. et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 28, 578–590 (2009).
Casares, N. et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 202, 1691–1701 (2005).
Spisek, R. et al. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood 109, 4839–4845 (2007).
Tesniere, A. et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 29, 482–491 (2010).
Yamazaki, T., Buque, A., Ames, T. D. & Galluzzi, L. PT-112 induces immunogenic cell death and synergizes with immune checkpoint blockers in mouse tumor models. Oncoimmunology 9, 1721810 (2020).
Senovilla, L. et al. An immunosurveillance mechanism controls cancer cell ploidy. Science 337, 1678–1684 (2012).
Nayagom, B. et al. Immunogenic cell death in a combined synergic gene- and immune-therapy against cancer. Oncoimmunology 8, e1667743 (2019).
Ocadlikova, D. et al. Chemotherapy-induced tumor cell death at the crossroads between immunogenicity and immunotolerance: focus on acute myeloid leukemia. Front. Oncol. 9, 1004 (2019).
Golden, E. B. et al. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology 3, e28518 (2014).
Gameiro, S. R. et al. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget 5, 403–416 (2014).
Garg, A. D., Krysko, D. V., Vandenabeele, P. & Agostinis, P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol. Immunother. 61, 215–221 (2012).
Li, W. et al. Targeting photodynamic and photothermal therapy to the endoplasmic reticulum enhances immunogenic cancer cell death. Nat. Commun. 10, 3349 (2019).
Tatsuno, K. et al. Extracorporeal photochemotherapy induces bona fide immunogenic cell death. Cell Death Dis. 10, 578 (2019).
Turubanova, V. D. et al. Immunogenic cell death induced by a new photodynamic therapy based on photosens and photodithazine. J. Immunother. Cancer 7, 350 (2019).
Gomes-da-Silva, L. C. et al. Photodynamic therapy with redaporfin targets the endoplasmic reticulum and Golgi apparatus. EMBO J. 37, e98354 (2018).
Adkins, I., Fucikova, J., Garg, A. D., Agostinis, P. & Spisek, R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology 3, e968434 (2014).
Fucikova, J. et al. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int J. Cancer 135, 1165–1177 (2014).
Nestvold, J. et al. Oncolytic peptide LTX-315 induces an immune-mediated abscopal effect in a rat sarcoma model. Oncoimmunology 6, e1338236 (2017).
Yamazaki, T. et al. The oncolytic peptide LTX-315 overcomes resistance of cancers to immunotherapy with CTLA4 checkpoint blockade. Cell Death Differ. 23, 1004–1015 (2016).
Xie, W. et al. Tumor lysis with LTX-401 creates anticancer immunity. Oncoimmunology 8, 1594555 (2019).
Zhou, H. et al. The oncolytic peptide LTX-315 triggers immunogenic cell death. Cell Death Dis. 7, e2134 (2016).
Zhou, H. et al. Oncolysis with DTT-205 and DTT-304 generates immunological memory in cured animals. Cell Death Dis. 9, 1086 (2018).
Bommareddy, P. K., Zloza, A., Rabkin, S. D. & Kaufman, H. L. Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma. Oncoimmunology 8, 1591875 (2019).
Pol, J. G. et al. Trial Watch: Oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology 7, e1503032 (2018).
Pol, J. et al. Trial Watch-Oncolytic viruses and cancer therapy. Oncoimmunology 5, e1117740 (2016).
Dudek, A. M., Garg, A. D., Krysko, D. V., De Ruysscher, D. & Agostinis, P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 24, 319–333 (2013).
Davola, M. E. & Mossman, K. L. Oncolytic viruses: how “lytic” must they be for therapeutic efficacy? Oncoimmunology 8, e1581528 (2019).
Ma, Y. et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 38, 729–741 (2013).
Sprooten, J., Agostinis, P. & Garg, A. D. Type I interferons and dendritic cells in cancer immunotherapy. Int. Rev. Cell Mol. Biol. 348, 217–262 (2019).
Gardai, S. J. et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334 (2005).
Poon, I. K., Lucas, C. D., Rossi, A. G. & Ravichandran, K. S. Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol. 14, 166–180 (2014).
Garg, A. D. et al. Resistance to anticancer vaccination effect is controlled by a cancer cell-autonomous phenotype that disrupts immunogenic phagocytic removal. Oncotarget 6, 26841–26860 (2015).
Krysko, D. V. et al. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 12, 860–875 (2012).
Vanpouille-Box, C., Hoffmann, J. A. & Galluzzi, L. Pharmacological modulation of nucleic acid sensors - therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 18, 845–867 (2019).
Michaud, M. et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577 (2011).
Apetoh, L. et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059 (2007).
Vacchelli, E. et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 350, 972–978 (2015).
Rodriguez-Ruiz, M. E. et al. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology 8, e1655964 (2019).
Zitvogel, L., Galluzzi, L., Kepp, O., Smyth, M. J. & Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 15, 405–414 (2015).
Sistigu, A. et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 20, 1301–1309 (2014).
Khodarev, N. N. Intracellular RNA sensing in mammalian cells: role in stress response and cancer therapies. Int. Rev. Cell Mol. Biol. 344, 31–89 (2019).
Garg, A. D. et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 31, 1062–1079 (2012).
Goicoechea, S., Orr, A. W., Pallero, M. A., Eggleton, P. & Murphy-Ullrich, J. E. Thrombospondin mediates focal adhesion disassembly through interactions with cell surface calreticulin. J. Biol. Chem. 275, 36358–36368 (2000).
Goicoechea, S., Pallero, M. A., Eggleton, P., Michalak, M. & Murphy-Ullrich, J. E. The anti-adhesive activity of thrombospondin is mediated by the N-terminal domain of cell surface calreticulin. J. Biol. Chem. 277, 37219–37228 (2002).
Eggleton, P. et al. Calreticulin is released from activated neutrophils and binds to C1q and mannan-binding protein. Clin. Immunol. Immunopathol. 72, 405–409 (1994).
Stuart, G. R., Lynch, N. J., Day, A. J., Schwaeble, W. J. & Sim, R. B. The C1q and collectin binding site within C1q receptor (cell surface calreticulin). Immunopharmacology 38, 73–80 (1997).
Ogden, C. A. et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 194, 781–795 (2001).
Verneret, M. et al. Relative contribution of c1q and apoptotic cell-surface calreticulin to macrophage phagocytosis. J. Innate Immun. 6, 426–434 (2014).
Segawa, K. et al. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344, 1164–1168 (2014).
Suzuki, J., Denning, D. P., Imanishi, E., Horvitz, H. R. & Nagata, S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406 (2013).
Nagata, S. Apoptosis and clearance of apoptotic cells. Annu. Rev. Immunol. 36, 489–517 (2018).
Buque, A., Rodriguez-Ruiz, M. E., Fucikova, J. & Galluzzi, L. Apoptotic caspases cut down the immunogenicity of radiation. Oncoimmunology 8, e1655364 (2019).
Fadok, V. A. et al. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 405, 85–90 (2000).
Li, M. O., Sarkisian, M. R., Mehal, W. Z., Rakic, P. & Flavell, R. A. Phosphatidylserine receptor is required for clearance of apoptotic cells. Science 302, 1560–1563 (2003).
Panaretakis, T. et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 15, 1499–1509 (2008).
Oldenborg, P. A., Gresham, H. D. & Lindberg, F. P. CD47-signal regulatory protein alpha (SIRPalpha) regulates Fcgamma and complement receptor-mediated phagocytosis. J. Exp. Med. 193, 855–862 (2001).
Iribarren, K. et al. Anticancer effects of anti-CD47 immunotherapy in vivo. Oncoimmunology 8, 1550619 (2019).
Chen, J. et al. Macrophages induce CD47 upregulation via IL-6 and correlate with poor survival in hepatocellular carcinoma patients. Oncoimmunology 8, e1652540 (2019).
Chao, M. P. et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med. 2, 63ra94 (2010).
Feng, M. et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 19, 568–586 (2019).
Truxova, I. et al. Calreticulin exposure on malignant blasts correlates with improved natural killer cell-mediated cytotoxicity in acute myeloid leukemia patients. Haematologica 105, 1868–1878 (2020).
Fucikova, J., Kline, J. P., Galluzzi, L. & Spisek, R. Calreticulin arms NK cells against leukemia. Oncoimmunology 9, 1671763 (2020).
Chen, X., Fosco, D., Kline, D. E. & Kline, J. Calreticulin promotes immunity and type I interferon-dependent survival in mice with acute myeloid leukemia. Oncoimmunology 6, e1278332 (2017).
Liu, P. et al. Immunosuppression by mutated calreticulin released from malignant cells. Mol. Cell 77, 748–760 (2020).
Alur, M. et al. Suppressive roles of calreticulin in prostate cancer growth and metastasis. Am. J. Pathol. 175, 882–890 (2009).
Peng, R. Q. et al. Expression of calreticulin is associated with infiltration of T-cells in stage IIIB colon cancer. World J. Gastroenterol. 16, 2428–2434 (2010).
Hsu, W. M. et al. Calreticulin expression in neuroblastoma–a novel independent prognostic factor. Ann. Oncol. 16, 314–321 (2005).
Fucikova, J. et al. Calreticulin expression in human non-small cell lung cancers correlates with increased accumulation of antitumor immune cells and favorable prognosis. Cancer Res. 76, 1746–1756 (2016).
Schardt, J. A., Weber, D., Eyholzer, M., Mueller, B. U. & Pabst, T. Activation of the unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin. Cancer Res. 15, 3834–3841 (2009).
Zhang, X. H. et al. Expression and significance of calreticulin in human osteosarcoma. Cancer Biomark. 18, 405–411 (2017).
Muth, C. et al. Primary glioblastoma multiforme tumors and recurrence: Comparative analysis of the danger signals HMGB1, HSP70, and calreticulin. Strahlenther. Onkol. 192, 146–155 (2016).
Kasikova, L. et al. Calreticulin exposure correlates with robust adaptive antitumor immunity and favorable prognosis in ovarian carcinoma patients. J. Immunother. Cancer 7, 312 (2019).
Fucikova, J. et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood 128, 3113–3124 (2016).
Wemeau, M. et al. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis. 1, e104 (2010).
Mendez-Ferrer, S. et al. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 20, 285–298 (2020).
Chen, C. N. et al. Identification of calreticulin as a prognosis marker and angiogenic regulator in human gastric cancer. Ann. Surg. Oncol. 16, 524–533 (2009).
Liu, R. et al. Calreticulin as a potential diagnostic biomarker for lung cancer. Cancer Immunol. Immunother. 61, 855–864 (2012).
Lwin, Z. M. et al. Clinicopathological significance of calreticulin in breast invasive ductal carcinoma. Mod. Pathol. 23, 1559–1566 (2010).
Eric, A. et al. Effects of humoral immunity and calreticulin overexpression on postoperative course in breast cancer. Pathol. Oncol. Res. 15, 89–90 (2009).
Matsukuma, S. et al. Calreticulin is highly expressed in pancreatic cancer stem-like cells. Cancer Sci. 107, 1599–1609 (2016).
Sheng, W. et al. Calreticulin promotes EGF-induced EMT in pancreatic cancer cells via Integrin/EGFR-ERK/MAPK signaling pathway. Cell Death Dis. 8, e3147 (2017).
Majeti, R. et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299 (2009).
Brightwell, R. M. et al. The CD47 “don’t eat me signal” is highly expressed in human ovarian cancer. Gynecol. Oncol. 143, 393–397 (2016).
Suzuki, S. et al. CD47 expression regulated by the miR-133a tumor suppressor is a novel prognostic marker in esophageal squamous cell carcinoma. Oncol. Rep. 28, 465–472 (2012).
Rumi, E. et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 123, 1544–1551 (2014).
Rotunno, G. et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 123, 1552–1555 (2014).
Tefferi, A. et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 124, 2507–2513 (2014).
Panagiota, V. et al. Prognostic effect of calreticulin mutations in patients with myelofibrosis after allogeneic hematopoietic stem cell transplantation. Leukemia 28, 1552–1555 (2014).
Palandri, F. et al. Differences in presenting features, outcome and prognostic models in patients with primary myelofibrosis and post-polycythemia vera and/or post-essential thrombocythemia myelofibrosis treated with ruxolitinib. New perspective of the MYSEC-PM in a large multicenter study. Semin. Hematol. 55, 248–255 (2018).
Ali, H. et al. MIPSS70+ v2.0 predicts long-term survival in myelofibrosis after allogeneic HCT with the Flu/Mel conditioning regimen. Blood Adv. 3, 83–95 (2019).
Kuykendall, A. T. et al. Genetically inspired prognostic scoring system (GIPSS) outperforms dynamic international prognostic scoring system (DIPSS) in myelofibrosis patients. Am. J. Hematol. 94, 87–92 (2019).
Hafner, A., Bulyk, M. L., Jambhekar, A. & Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 20, 199–210 (2019).
Fang, D. D. et al. MDM2 inhibitor APG-115 synergizes with PD-1 blockade through enhancing antitumor immunity in the tumor microenvironment. J. Immunother. Cancer 7, 327 (2019).
Perdrix, A. et al. PRIMA-1 and PRIMA-1(Met) (APR-246): From mutant/wild type p53 reactivation to unexpected mechanisms underlying their potent anti-tumor effect in combinatorial therapies. Cancers 9, 172 (2017).
Pronier, E. et al. Targeting the CALR interactome in myeloproliferative neoplasms. JCI Insight 3, e122703 (2018).
Galluzzi, L., Bravo-San Pedro, J. M., Levine, B., Green, D. R. & Kroemer, G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 16, 487–511 (2017).
Hu, Z., Ott, P. A. & Wu, C. J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 18, 168–182 (2018).
Kaplan, D. H. et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 95, 7556–7561 (1998).
Mumberg, D. et al. CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proc. Natl. Acad. Sci. USA 96, 8633–8638 (1999).
Shankaran, V. et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410, 1107–1111 (2001).
Dunn, G. P., Old, L. J. & Schreiber, R. D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 22, 329–360 (2004).
Vesely, M. D., Kershaw, M. H., Schreiber, R. D. & Smyth, M. J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 29, 235–271 (2011).
Martinek, J., Wu, T. C., Cadena, D., Banchereau, J. & Palucka, K. Interplay between dendritic cells and cancer cells. Int. Rev. Cell Mol. Biol. 348, 179–215 (2019).
Lee, Y. S. & Radford, K. J. The role of dendritic cells in cancer. Int. Rev. Cell Mol. Biol. 348, 123–178 (2019).
Balan, S., Saxena, M. & Bhardwaj, N. Dendritic cell subsets and locations. Int. Rev. Cell Mol. Biol. 348, 1–68 (2019).
Rao, S., Gharib, K. & Han, A. Cancer immunosurveillance by T cells. Int. Rev. Cell Mol. Biol. 342, 149–173 (2019).
Martinez, R. et al. Combined assessment of peritumoral Th1/Th2 polarization and peripheral immunity as a new biomarker in the prediction of BCG response in patients with high-risk NMIBC. Oncoimmunology 8, 1602460 (2019).
Li, Z. et al. Genomic and transcriptional profiling of tumor infiltrated CD8(+) T cells revealed functional heterogeneity of antitumor immunity in hepatocellular carcinoma. Oncoimmunology 8, e1538436 (2019).
Lopez-Soto, A., Gonzalez, S., Smyth, M. J. & Galluzzi, L. Control of metastasis by NK cells. Cancer Cell 32, 135–154 (2017).
Chiossone, L., Dumas, P. Y., Vienne, M. & Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 18, 671–688 (2018).
Cursons, J. et al. A gene signature predicting natural killer cell infiltration and improved survival in melanoma patients. Cancer Immunol. Res. 7, 1162–1174 (2019).
Wculek, S. K. et al. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20, 7–24 (2020).
Borst, J., Ahrends, T., Babala, N., Melief, C. J. M. & Kastenmuller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 18, 635–647 (2018).
Yarchoan, M., Johnson, B. A. 3rd, Lutz, E. R., Laheru, D. A. & Jaffee, E. M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 17, 209–222 (2017).
Amon, L. et al. Transcriptional control of dendritic cell development and functions. Int. Rev. Cell Mol. Biol. 349, 55–151 (2019).
Picard, E. et al. Circulating NKp46(+) natural killer cells have a potential regulatory property and predict distinct survival in non-small cell lung cancer. Oncoimmunology 8, e1527498 (2019).
Zhang, H. et al. The role of NK cells and CD39 in the immunological control of tumor metastases. Oncoimmunology 8, e1593809 (2019).
Shaul, M. E. & Fridlender, Z. G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 16, 601–620 (2019).
Prendergast, G. C., Malachowski, W. J., Mondal, A., Scherle, P. & Muller, A. J. Indoleamine 2,3-dioxygenase and its therapeutic inhibition in cancer. Int. Rev. Cell Mol. Biol. 336, 175–203 (2018).
Vitale, I., Manic, G., Coussens, L. M., Kroemer, G. & Galluzzi, L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 30, 36–50 (2019).
Cassetta, L. & Pollard, J. W. Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17, 887–904 (2018).
Guerriero, J. L. Macrophages: their untold story in T cell activation and function. Int. Rev. Cell Mol. Biol. 342, 73–93 (2019).
Petitprez, F. et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560 (2020).
Dasgupta, S., Dasgupta, S. & Bandyopadhyay, M. Regulatory B cells in infection, inflammation, and autoimmunity. Cell Immunol. 352, 104076 (2020).
Leylek, R. & Idoyaga, J. The versatile plasmacytoid dendritic cell: function, heterogeneity, and plasticity. Int. Rev. Cell Mol. Biol. 349, 177–211 (2019).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541 (2018).
Brandizzi, F. & Barlowe, C. Organization of the ER-Golgi interface for membrane traffic control. Nat. Rev. Mol. Cell Biol. 14, 382–392 (2013).
Galluzzi, L. & Green, D. R. Autophagy-independent functions of the autophagy machinery. Cell 177, 1682–1699 (2019).
Acknowledgements
G.K. is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) — Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; High-end Foreign Expert Program in China (GDW20171100085 and GDW20181100051), Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM). L.G. is supported by a Breakthrough Level 2 grant from the US Department of Defense (DoD), Breast Cancer Research Program (BRCP) (#BC180476P1), by the 2019 Laura Ziskin Prize in Translational Research (#ZP-6177, PI: Formenti) from the Stand Up to Cancer (SU2C), by a Mantle Cell Lymphoma Research Initiative (MCL-RI, PI: Chen-Kiang) grant from the Leukemia and Lymphoma Society (LLS), by a startup grant from the Department of Radiation Oncology at Weill Cornell Medicine (New York, USA), by a Rapid Response Grant from the Functional Genomics Initiative (New York, USA), by industrial collaborations with Lytix (Oslo, Norway) and Phosplatin (New York, USA), and by donations from Phosplatin (New York, US), the Luke Heller TECPR2 Foundation (Boston, USA) and Sotio a.s. (Prague, Czech Republic).
Author information
Authors and Affiliations
Contributions
J.F. and L.G. conceived the manuscript. J.F. and L.G. wrote the first version of the manuscript with constructive input from R.S. and G.K. J.F. prepared display items under supervision from L.G. L.G. addressed issues raised by reviewers and requested by editors. All authors approved the final version of the article.
Corresponding authors
Ethics declarations
Competing interests
J.F. and R.S. are full-time employees of Sotio. G.K. has been holding research contracts with Bayer Healthcare, Genentech, Glaxo Smyth Kline, Institut Mérieux, Lytix Pharma, PharmaMar, Sotio and Vasculox. He is on the Board of Directors of the Bristol Myers Squibb Foundation France, member of the Scientific Advisory Board of The Longevity Labs, and scientific co-founder of everImmune, Samsara therapeutics and Therafast Bio. L.G. received research support from Lytix and Phosplatin, consulting fees from OmniSEQ, Astra Zeneca, Inzen and the Luke Heller TECPR2 Foundation, and he is member of Scientific Advisory Committees for Boehringer Ingelheim, The Longevity Labs and OmniSEQ.
Rights and permissions
About this article

Cite this article
Fucikova, J., Spisek, R., Kroemer, G. et al. Calreticulin and cancer. Cell Res 31, 5–16 (2021). https://doi.org/10.1038/s41422-020-0383-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-020-0383-9
This article is cited by
-
Cell death by phagocytosis
Nature Reviews Immunology (2024)
-
A novel anoikis-related gene signature identifies LYPD1 as a novel therapy target for bladder cancer
Scientific Reports (2024)
-
DNA polymerase beta connects tumorigenicity with the circadian clock in liver cancer through the epigenetic demethylation of Per1
Cell Death & Disease (2024)
-
Targeting immunogenic cell stress and death for cancer therapy
Nature Reviews Drug Discovery (2024)
-
Immunogenic Cell Death-related Signature Evaluates the Tumor Microenvironment and Predicts the Prognosis in Diffuse Large B-Cell Lymphoma
Biochemical Genetics (2024)
