
Abstract
In the present study, to improve the predictive performance of a model and its reproducibility when applied to an independent data set, we investigated the use of multimodel inference to predict the probability of having a complex psychiatric disorder. We formed training and test sets using proteomic data (147 peptides from 77 proteins) from two-independent collections of first-onset drug-naive schizophrenia patients and controls. A set of prediction models was produced by applying lasso regression with repeated tenfold cross-validation to the training set. We used feature extraction and model averaging across the set of models to form two prediction models. The resulting models clearly demonstrated the utility of a multimodel based approach to make good (training set AUC > 0.80) and reproducible predictions (test set AUC > 0.80) for the probability of having schizophrenia. Moreover, we identified four proteins (five peptides) whose effect on the probability of having schizophrenia was modified by sex, one of which was a novel potential biomarker of schizophrenia, foetal haemoglobin. The evidence of effect modification suggests that future schizophrenia studies should be conducted in males and females separately. Future biomarker studies should consider adopting a multimodel approach and going beyond the main effects of features.
Similar content being viewed by others


Introduction
Despite our ever increasing ability to generate data, many published findings are not reproducible in independent data sets1,2,3. In biological psychiatry, this situation is further exaggerated by the lack of a biological ‘gold standard’ diagnoses for psychiatric disorders4,5, which are still diagnosed based on the evaluation of signs and symptoms in clinical interviews. One notable limitation of symptom-based diagnosis is that the boundaries between disorders can be poorly defined because of overlapping symptoms and common co-morbidity across psychiatric disorders, which can result in a layering or commingling of symptoms6. Consequently, patient groups are biologically heterogenous4, misdiagnosis is common7 and prediction models attempt to link biological data to a symptom-based diagnosis4,5.
Nevertheless, although many significant psychiatric disorder biomarker findings have been reported, only few have been consistently replicated8,9. This lack of reproducibility is a result of underpowered replication studies for the small to moderate effect sizes initially reported3,5, differences in patient selection criteria5 (e.g. age, recency of diagnosis, sex ratio10, treatment, and comorbidities) and inconsistencies in methods used to quantify biological markers. In addition, when model selection is performed in high-dimensional data, defined as data with more features (variables) than subjects, overfitting a model can be a major issue and be compounded by biologically heterogeneous psychiatric patient populations. Overfitting occurs when the coefficient estimates of the selected model depend not only on the underlying relationship of interest, but also on chance characteristics of the data analysed. When an overfitted model is applied to new subjects the predictive performance is reduced. In other words, the model provides an over-optimistic assessment of the predictive performance when based on the data to which the model was fitted.
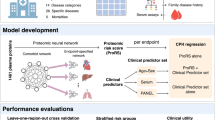
Despite the heterogeneity of psychiatric patients, models with a more reproducible predictive performance can be achieved by taking into account model selection uncertainty. Model selection is often considered to be a process of selecting a single model from a set of all possible models that is judged to be the ‘best’ model for making inferences from the analysed data11,12. Any uncertainty in model selection, for example, resulting from a small change in the data set, is ignored once the best model has been found. In the present study, rather than making predictions based on a single-best model selected from a data set, we adopted a multimodel approach to make predictions for the probability of having schizophrenia based on a set of models to allow for any uncertainty in model selection. The schizophrenia data analysed consisted of two-independent mass spectrometry (MS) multiple reaction monitoring (MRM) proteomic data sets (147 peptides from 77 proteins) of first-onset drug-naive schizophrenia patients and controls13,14 that were used in the present study as training and test sets. The sensitivity of model selection to small changes in the training data was evaluated by model selection using least absolute shrinkage and selection operator (lasso) regression with the resampling approach of repeated tenfold cross-validation15. In the absence of one model being superior to the other selected models, we used feature extraction and model averaging across the set of models to form the prediction model. This approach improved the generalizability of the model, that is, it reduced overfitting and provided more reproducible inference. We then attempted to validate the model predictive performance by applying the model to an independent test set.
Materials and methods
Subjects
The Cologne study (referred to as the ‘training set’), as previously described13, consisted of serum samples from 60 first-onset drug-naive schizophrenia patients and 79 age and sex matched controls recruited by the Department of Psychiatry, University of Cologne (Supplementary Table 1). The Rotterdam study (referred to as the ‘independent test set’), as previously described14, consisted of nine first-onset drug-naive male schizophrenia patients and 12 male controls recruited by the Erasmus Medical Centre in Rotterdam (Supplementary Table 1). Schizophrenia was diagnosed based on the Diagnostic and Statistical Manual of Mental Disorders IV (DSM-IV)16. The ethical committees of the medical faculties of the respective universities approved the protocols of the study. Informed consent was given in writing by all participants and clinical investigations were conducted according to the Declaration of Helsinki.
Targeted protein quantification
Serum samples were prepared in a 96-well plate format as described previously17. Briefly, serum samples were diluted with ammonium bicarbonate. Then, disulphide bond reduction and cysteine alkylation were performed using Dithiothreitol (DTT) and Iodoacetamide (IAA), respectively. Proteins were digested overnight using trypsin (see Supplementary Information). Isotopically labelled internal standard peptides were spiked into serum samples prior to MS run. Quality control (QC) samples were used in this study to monitor method performance and instrument stability (see Supplementary Information).
In this study, a total of 101 proteins (172 peptides), the majority previously associated with psychiatric disorders, were selected. Three to four interference free transitions were selected for each targeted peptide as described previously17. Tryptic digested peptides were monitored using an Agilent 1290 liquid chromatography (LC) system coupled with 6495 Triple Quadrupole mass spectrometer equipped with jet-stream nano ESI source operated in positive mode. MS data were acquired in MRM mode. The chromatographic separation was carried out on Agilent AdvanceBio Peptide Map column (2.1 × 150 mm 2.7-micron) at 50 °C. Peptides were eluted over a linear gradient from 3 to 30% acetonitrile in 0.1% formic acid in 45 min.
Statistical analysis
Data pre-processing and quality control
We processed raw mass spectrometry (MS) files using the Skyline software package (Version 3.1.0)18. We manually checked peaks and when necessary, adjusted peak integrations accordingly. The endogenous and internal standard peptide-transition peak areas were estimated and exported as a comma delimited data file for statistical analysis in R (Version 3.4.4)19. The MS data pre-processing is described in Supplementary Information.
Model selection
We used lasso regression with repeated tenfold cross-validation to reduce overfitting and to investigate model selection uncertainty in the training set.
Tenfold cross-validation
Tenfold cross-validation is a commonly used resampling approach to reduce the problem of overfitting11. The data are randomly split into tenfolds. We hold out each fold one at a time, train on the remaining data and predict the held out observations for each value of the regularization parameter – selecting the regularization parameter that minimises the cross-validation deviance (classification error). The model, as defined by the regularization parameter, is then fit to the entire data set11. We repeatedly applied tenfold cross-validation 100 times to determine how sensitive model selection was to small changes in the training set (overfitting). Note that changes in the training set result from the data being randomly split into tenfolds for each application of tenfold cross-validation.
Lasso regression
Lasso regression is a penalized regression approach that reduces overfitting by placing a constraint on the sum of the absolute values of the regression coefficients, which shrinks the coefficients, a process referred to as regularization or shrinkage, and allows poor predictors to be shrunken exactly to zero (variable selection)20. Shrinkage often improves the prediction accuracy11. The constraint (also known as the regularization parameter, shrinkage parameter or penalty) was selected using tenfold cross-validation. Lasso regression with tenfold cross-validation was conducted using the R package glmnet15,20. We set the elastic-net penalty, α, that bridges the gap between lasso (α = 1, the default) and ridge regression (α = 0), to 0.9 for numerical stability15,20.
Akaike information criterion
We adopted a model averaging approach using the Akaike information criterion (AIC) weights as described in Burnham and Anderson11,12. We calculated the AIC for each model selected by lasso regression with tenfold cross-validation. The AIC is a measure of how well a model fits the data relative to the other possible models given the data analysed and favours fewer parameters21. The model with the lowest AIC is the best model approximating the outcome of interest. AIC can be expressed as:
where K = number of model parameters and log-likelihood is a measure of model fit12. In this study, as n/K ≤ 40 for sample size n and the model with the largest value of K, we used the second-order bias correction version of the AIC:
where n = sample size, K = number of model parameters and log-likelihood is a measure of model fit12.
Akaike weights
After model selection, we calculated the Akaike weights, wm, for each model:
where wm and AICm are, respectively, the Akaike weight and AICc for model m and AICj is the AICc for model j = 1 to M. The denominator normalizes the Akaike weights, so that
The Akaike weights can be interpreted as model probabilities or the ‘weight of evidence’ in favour of model m being the best model based on the available data set12. The Akaike weights can be used to quantify the evidence for the importance of each feature in the set of selected models11. The relative importance of feature f is the summation of the Akaike weights across the set of selected models which include feature f11. The resulting relative feature importance, between 0 and 1, allows features to be ranked by their importance and can be interpreted as the probability of the feature being included in the best model for the data. The relative importance of subsets of features occurring together can also be quantified11.
Feature extraction
We defined two prediction models, the first based the frequency of feature selection (inclusion fraction > 0.80) and the second based on the probability that a feature is included in the best model for the data (inclusion probability > 0.90). Both models have the added advantage of further reducing overfitting by excluding features less frequently selected or with low inclusion probabilities (Akaike weights). We note that both the inclusion fraction and probability thresholds were determined before the analysis was conducted.
Model averaging
If one of the selected models was clearly superior to the other selected models [for example, it has a probability that it is the best model for the data, w > 0.911], then inference could be based on that model alone. However, when model selection uncertainty is evident, inference based on a set of models can result in more reproducible inference. To obtain more reproducible predictions of schizophrenia diagnosis, we adopted a model averaging approach, specifically, for a feature of interest, we estimated their weighted average coefficient, \(\widehat {\bar \beta }\), across a set of models derived from applying lasso regression with repeated tenfold cross-validation to the training data set.
\(\widehat {\bar \beta } = \mathop {\sum }\limits_{j = 1}^M w_j\hat \beta _j\), where wj and \(\hat \beta _j\) are, respectively, the Akaike weight and coefficient estimates for a feature of interest in model j and \(\widehat {\bar \beta }\) is the weighted average of \(\hat \beta _j\) over models j = 1 to M11. As all models are considered, those models not containing the feature of interest contribute zero to the weighted average estimate which results in the coefficient shrinking towards zero.
Model selection including first-order interactions with sex
As the two prediction models had a substantially better predictive performance for males than for females (Table 2a), we wanted to investigate whether the effect of protein abundance on the probability of having schizophrenia was modified by the sex of the individual. To investigate effect modification, we needed to consider first-order interactions between protein abundance and sex in model selection. If we consider a simple logistic model
where Pr(Y = 1) = π, logit(π) is the logit function of this probability [natural log of π/(1 – π)], α is the intercept and, β1 and β2 are coefficients for features X1 and X2. Typically, a product term X1X2 is added to the model to allow for an interaction
The coefficient of the product term, β3, reflects interaction as a departure from the multiplicative effects, in other words, the combined effect of X1 and X2 is larger (or smaller) than the product of their individual main effects.
To consider interactions between protein abundance and sex in the model selection with repeated tenfold cross-validation, we adopted the glinternet approach of Lim and Hastie22. Glinternet is based on group lasso23 and importantly, ensures hierarchically well formulated models, that is, an interaction can only be selected if both of its main effects are selected22. In other words, if an interaction term is selected and main effects have not, the main effects will also be selected. We used the R package glinternet22.
Predictive performance
Predictive performance was evaluated using the area under the receiver operating characteristic (ROC) curve. The area under the curve (AUC) measures the extent to which a model’s predicted probability agrees with the observed outcome, that is the presence or absence of an event. The AUC is the probability that a randomly chosen patient with the event is rated/ranked higher than a randomly chosen patient without the event. A model performing no better than random will have an AUC of 0.50. (AUC: 0.9–1 = excellent; 0.8–0.9 = good; 0.7–0.8 = fair; 0.6–0.7 = poor; 0.5–0.6 = fail). The AUC was calculated using the R package ROCR24.
Pathway analysis
Biological process pathway analysis was carried out using Gene Ontology and PANTHER25. UniProt accession numbers of proteins corresponding to the peptides selected in the final model were uploaded to http://geneontology.org and all Homo sapiens genes in the database were used as a reference list. Fisher’s exact with false discovery rate (FDR) multiple test correction was used for determining pathway significance.
Results
After MS data pre-processing, there were 77 proteins (147 peptides) measured in a training set of 60 first-onset drug-naive schizophrenia patients and 77 controls, and in a male-only independent test set of nine first-onset drug-naive schizophrenia patients and 12 controls (Supplementary Table 1).
Model selection
Model selection using lasso regression with repeated tenfold cross-validation revealed some uncertainty in model selection in the training set (Fig. 1). Based on the features selected, there were 11 unique models (Supplementary Table 2) with, on average, a good predictive performance (AUC = 0.81; Fig. 1a). The number of features selected ranged from 3 to 33, with eight on average (Fig. 1b). Although 33 features out of 149 (147 peptides, sex, and age) were selected at least once (Fig. 1c; Supplementary Table 3), 25 of these features were selected less than 10 times out of 100, consistent with limited model overfitting. The remaining eight features were selected more than 50 times, six of which were selected more than 80 times out of 100 (Table 1). There was not a single best model approximating schizophrenia status in the training set; the most frequently selected model had eight features and was selected 51 times out of 100.

a A bar chart summarizing the AUCs for each model. AUC: Fail 0.5–0.6; Poor 0.6–0.7; Fair 0.7–0.8; Good 0.8–0.9; and, 0.9–1.00 Excellent. b A bar chart summarizing the number of features selected in each model. c An inclusion fraction plot summarizing the proportion of times each feature was selected in a model. One hundred and seventeen features out of 149 (147 peptides, sex, and age) were not selected in the 100 models. d A plot of inclusion frequencies and probabilities for the 32 selected features
Akaike model weights
Akaike weights were calculated for each of the 100 selected models. As there were 11 unique models, we summed the weights by each unique model to estimate the probability of it being the best model approximating the probability of having schizophrenia in the training set. Despite some uncertainty in model selection, the three models with the highest probability of being the best model had a combined probability of over 0.95; five models had a combined probability of over 0.99 (Supplementary Table 2). The most frequently selected model had the highest single model probability of 0.80. We note that frequency of selection does not necessarily equate to the probability of being the best model for the data analysed (Figs 1d and 2f; Supplementary Tables 1 and 3).

a A bar chart summarizing the AUCs for each model. AUC: Fail 0.5–0.6; Poor 0.6–0.7; Fair 0.7–0.8; Good 0.8–0.9; and, 0.9–1.00 Excellent. b A bar chart summarizing the number of features selected in each model. c A bar chart summarizing the number of first-order interactions with sex selected in each model. d An inclusion fraction plot summarizing the proportion of times each feature was selected in a model. e An inclusion fraction plot summarizing the proportion of times each first-order interactions with sex was selected in a model. One hundred and twenty six features out of 149 (147 peptides, sex, and age) were not selected in the 100 models. f A plot of inclusion frequencies and probabilities for the selected features and interactions
Model averaging
We defined two models using feature extraction (Table 1), the first consisting of six features with an inclusion fraction >0.8 and the second consisting of eight features with an inclusion probability (relative feature importance) >0.9. Model averaging for each feature of interest was conducted across all 100 selected models. After model averaging, the first model (six features) was applied to the training set and then to the independent test set, both had a good predictive performance, AUC of 0.81 and 0.88, respectively (Table 2a). A similar predictive performance was obtained when the second model (eight features) was applied to the training set and then to the independent test set, AUC of 0.82 and 0.92, respectively (Table 2a).
To investigate the higher predictive performance in the male-only independent test set, we then applied the two models to training set males and females separately. We found that the predictive performance was substantially higher for males than for females (Table 2a). We note that sex had not been selected in any of the 100 models (Supplementary Tables 1 and 2) and that first-order interactions between features and sex had not been considered in the model selection.
Model selection considering first-order interactions with sex
To investigate potential differences between males and females, we adopted the glinternet approach of Lim and Hastie22 to consider first-order interactions between protein abundance and sex in model selection with repeated tenfold cross-validation. Importantly, this approach ensures that the model obeys strong hierarchy, that is, if an interaction is selected, both of its main effects will also be selected22,26. We only considered first-order interactions with sex.
After considering interactions in model selection, there was less uncertainty in model selection. Based on the features selected, there were five unique models (Supplementary Table 4) with, on average, a good predictive performance (AUC = 0.87; Fig. 2a). Notably, all five of the unique models included interaction terms. The vast majority of selected features (19/23; Fig. 2d) and interactions (5/7; Fig. 2e) were selected at least 98 times out of 100. The two APOC3 peptides were selected in every model despite a strong linear relationship between them (correlation coefficient r = 0.87 and P < 2.2 × 10−16). The eight features selected in the earlier analysis (Table 1) were selected in every model, but none had interactions with sex selected (Supplementary Table 4). The five frequently selected interactions suggest that the sex of a subject was modifying the effects of peptides from APOE, A2AP, HBA, HBG1, and SHBG on the probability of having schizophrenia. The difference between males and females in the abundance of these five peptides in schizophrenia patients and controls is shown in Supplementary Figure 3. Consequently, model selection should be conducted in males and females separately to allow for biological differences between the sexes. However, in the present study, the training set was too small to conduct model selection in males and females separately.
As in the previous analysis, we defined two models (Table 3), the first consisting of 17 features and five interactions with an inclusion fraction of >0.8 and the second consisting of 13 features and three interactions with an inclusion probability >0.9. Model averaging for each feature of interest was conducted across all 100 selected models. After model averaging, the first model was applied to the training set and then to the independent test set. The marked difference seen in the previous analysis was no longer evident and both had a good predictive performance, AUC of 0.86 and 0.89, respectively, demonstrating the reproducibility of the model (Table 2b). A similar predictive performance was obtained when the second model was applied to the training set and then to the independent test set, AUC of 0.85 and 0.82, respectively (Table 2b). Despite the good predictive performance and the reproducibility, when we applied the two models to training set males and females separately, although marginally less than in the previous analysis, the predictive performance remained substantially better for males than for females (Table 2b).
Discussion
Seventy-seven proteins (147 peptides) were measured in serum samples from two-independent collections of first-onset drug-naive schizophrenia patients and matched controls using mass spectrometry. We were able to demonstrate that multimodel inference can provide models for the probability of having schizophrenia with a good (training set AUC > 0.8) and reproducible predictive performance (test set AUC > 0.8; Table 2). A notable advantage of the multimodel approach, particularly evident from the set of selected models (Supplementary Tables 1 and 3), is that it reduces the risk of selecting one of the less probable models by chance. In addition, feature extraction using the inclusion fractions or probabilities to select features for the model has the effect of further limiting model overfitting. Although we used both inclusion fractions and probabilities for feature selection, the latter is a more sensible approach as it represents how likely each feature is to be included in the best model for the data analysed.
After observing the differences in the prediction performance between males and females in the training set (Table 2a), extending the analysis to include first-order interactions was essential to determine whether the effect of protein abundance on the probability of having schizophrenia was modified by sex. As the vast majority of the selected models contained the same five interactions (Supplementary Table 4), there was sufficient evidence of modification by sex, suggesting that future schizophrenia biomarker studies should ideally be conducted in males and females separately. This would allow for biological differences underpinning the reported sex differences in schizophrenia to be better utilized in the prediction model. Reported sex differences include males having an earlier onset, more negative and less depressive symptoms while females experience more emotional and psychotic symptoms28,29.
Despite concerns about the impact of symptom-based diagnosis of schizophrenia on model selection uncertainty, resampling using repeated tenfold cross-validation revealed a large degree of stability in the features selected (Table 2; Fig. 1c) that was notably improved with the inclusion of first-order interactions with sex (Table 3; Fig. 2d, e). The similar participant selection criteria, particularly the recruitment of first-onset drug-naive schizophrenia patients, in the training and independent test sets would have contributed to this stability (Supplementary Table 1; see Methods).
As 30 of the 77 proteins analysed have been previously associated with schizophrenia (Supplementary Table 6), it was re-assuring, although not unexpected, that the majority of the selected proteins (12 of 14; Table 3) have been previously associated with schizophrenia30,31,32,33 with the most robust finding for increased HPT levels in schizophrenia patients compared to controls30,34,35,36,37. In addition, the most significant pathways related to the selected proteins, namely altered acute inflammatory response [involving ANT3, APOA2, A2AP, FETUA, HPT, and ITIH4; gene ontology (GO) term GO:0002526, false discovery rate (FDR) = 1.6 × 10–7] and ultra-low density lipoprotein transport (APOA2, APOA4, APOC3 and APOE; GO:0034378, GO:0034371 and GO:0034370, FDR < 1.0 × 10−6) in schizophrenia, are consistent with previous reports36,38. The fact that proteins from these pathways also correlate with disease psychopathology scores, in particular-negative symptoms39,40,41, suggests that these processes might mechanistically underpin certain aspects of schizophrenia pathophysiology and as such constitute promising drug targets for add-on treatments.
Model selection using glinternet to allow for first-order interactions identified five peptides from APOE, A2AP, HBA, HBG1 and SHBG (Table 3) whose effect on the probability of having schizophrenia was modified by sex. Interestingly, neither sex nor any of the five protein peptides were selected in the initial model selections that did not consider interactions (Supplementary Table 3). While APOE, A2AP, and SHBG have been previously implicated in schizophrenia33,42,43, the association with HBA and HBG1 has not been reported before. HBA and HBG1 are haemoglobin subunits alpha and gamma-1, respectively. Although subunit alpha may belong to different types of haemoglobin (e.g. A, A2 and F), the selection of peptides from subunits alpha and gamma-1 by the algorithm, with the latter subunit specific to foetal haemoglobin (haemoglobin F or haemoglobin α2γ2), as well as a moderate linear relationship between the subunits (r = 0.57 and P = 2.0 × 10−7), indicate that this findings may represent foetal haemoglobin44.
There are several limitations to the present study. First, the independent training and test sets had a relatively small sample size with limited demographic and clinical data measured. Second, although a male-only independent test set was not ideal, it proved to be insightful with regard to the sex differences in the protein effects on the probability of having schizophrenia. Had the independent test set consisted of males and females, the reproducibility of the prediction model would have depended on how close the sex ratio was between the two data sets and the sex differences would have been less evident. Finally, we have fitted models to distinguish patients from controls, although this may not reflect the relevant clinical population for a schizophrenia diagnostic test, it is an appropriate first step towards distinguishing between individuals at an ultra-high risk of developing schizophrenia who do and do not develop the disorder over a set time period.
In conclusion, we have demonstrated the utility of a multimodel-based approach to make good and reproducible predictions for a complex psychiatric disorder. We have also demonstrated the importance of considering first-order interactions in model selection and propose that future biomarker studies of schizophrenia should be conducted in males and females separately.
Data availability
The data sets analysed during the current study are available from the corresponding author on reasonable request.
References
Baker, M. Is there a reproducibility crisis? Nature 533, 452–454 (2016).
Yang, Y. & Zou, H. A fast unified algorithm for solving group-lasso penalize learning problems. Stat. Comput. 1129–1141 (2014).
Ioannidis, J. P. Why most published research findings are false. PLoS Med. 2, e124 (2005).
Insel, T. R. The NIMH Research Domain Criteria (RDoC) Project: precision medicine for psychiatry. Am. J. Psychiatry 171, 395–397 (2014).
Kapur, S., Phillips, A. G. & Insel, T. R. Why has it taken so long for biological psychiatry to develop clinical tests and what to do about it? Mol. Psychiatry 17, 1174–1179 (2012).
Federman, R. Misdiagnosis of Bipolar Disorder, https://www.psychologytoday.com/intl/blog/bipolar-you/201402/misdiagnosis-bipolar-disorder
Gonzalez-Pinto, A. et al. First episode in bipolar disorder: misdiagnosis and psychotic symptoms. J. Affect Disord. 50, 41–44 (1998).
Mischak, H. et al. Implementation of proteomic biomarkers: making it work. Eur. J. Clin. Invest. 42, 1027–1036 (2012).
Niculescu, A. B. et al. Psychiatric blood biomarkers: avoiding jumping to premature negative or positive conclusions. Mol. Psychiatry 20, 286–288 (2015).
Ramsey, J. M., Cooper, J. D., Penninx, B. W. & Bahn, S. Variation in serum biomarkers with sex and female hormonal status: implications for clinical tests. Sci. Rep. 6, 26947 (2016).
Burnham, K. P. & Anderson, D. R. Model Selection and Multimodel Inference 2nd edn, (Springer, New York, 2002).
Burnham, K. P. & Anderson, D. R. Multimodel inference: understanding AIC and BIC in model selection. Sociol. Methods Res. 33, 261–304 (2004).
Cooper, J. D. et al. Schizophrenia-risk and urban birth are associated with proteomic changes in neonatal dried blood spots. Transl. Psychiatry 7, 1290 (2017).
Tomasik, J. et al. Pretreatment levels of the fatty acid handling proteins H-FABP and CD36 predict response to olanzapine in recent-onset schizophrenia patients. Brain Behav. Immunol. 52, 178–186 (2016).
Hastie, T., Tibshirani, R. & Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction 5th edn (Springer, New York, NY, USA, 2001).
American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-IV 4th edn, (American Psychiatric Association, Washington, 1994).
Ozcan, S. et al. Towards reproducible MRM based biomarker discovery using dried blood spots. Sci. Rep. 7, 45178 (2017).
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
R. Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing (Vienna, Austria, 2015).
Tibshirani, R. Regression shrinkage and selection via the Lasso. J. Roy. Stat. Soc. B Met 58, 267–288 (1996).
Akaike, H. Information theory as anextension of the maximum likelihood principle, 267–281 (Budapest, Akaemiai Kiado, 1973).
Lim, M. & Hastie, T. Learning interactions via hierarchical group-lasso regularization. J. Comput. Graph Stat. 24, 627–654 (2015).
Yuan, M. & Lin, Y. Model selection and estimation in regression with grouped variables. J. Roy. Stat. Soc. B 68, 49–67 (2006).
Sing, T., Sander, O., Beerenwinkel, N. & Lengauer, T. ROCR: visualizing classifier performance in R. Bioinformatics 21, 3940–3941 (2005).
Mi, H. et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45, D183–D189 (2017).
Jaccard, J. Interaction effects in logistic regression (Sage Publications, Thousand Oaks, California, 2001).
The UniProt, C.. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017).
Castle, D. J. & Buckley, P. F. Schizophrenia 2nd edn, (Oxford University Press, Oxford, 2015).
Abel, K. M., Drake, R. & Goldstein, J. M. Sex differences in schizophrenia. Int. Rev. Psychiatry 22, 417–428 (2010).
Chan, M. K. et al. Development of a blood-based molecular biomarker test for identification of schizophrenia before disease onset. Transl. Psychiatry 5, e601 (2015).
Levin, Y. et al. Global proteomic profiling reveals altered proteomic signature in schizophrenia serum. Mol. Psychiatry 15, 1088–1100 (2010).
Domenici, E. et al. Plasma protein biomarkers for depression and schizophrenia by multi analyte profiling of case-control collections. PLoS ONE 5, e9166 (2010).
Ramsey, J. M. et al. Distinct molecular phenotypes in male and female schizophrenia patients. PLoS ONE 8, e78729 (2013).
Wan, C. et al. Abnormal changes of plasma acute phase proteins in schizophrenia and the relation between schizophrenia and haptoglobin (Hp) gene. Amino Acids 32, 101–108 (2007).
Yang, Y. et al. Altered levels of acute phase proteins in the plasma of patients with schizophrenia. Anal. Chem. 78, 3571–3576 (2006).
Maes, M. et al. Acute phase proteins in schizophrenia, mania and major depression: modulation by psychotropic drugs. Psychiatry Res. 66, 1–11 (1997).
Schwarz, E. et al. Validation of a blood-based laboratory test to aid in the confirmation of a diagnosis of schizophrenia. Biomark. Insights 5, 39–47 (2010).
Pillinger, T., Beck, K., Stubbs, B. & Howes, O. D. Cholesterol and triglyceride levels in first-episode psychosis: systematic review and meta-analysis. Br. J. Psychiatry 211, 339–349 (2017).
Morera, A. L., Henry, M., Garcia-Hernandez, A. & Fernandez-Lopez, L. Acute phase proteins as biological markers of negative psychopathology in paranoid schizophrenia. Actas Esp. Psiquiatr. 35, 249–252 (2007).
Gohar, S. M. et al. Association between serum lipid levels, osteoprotegerin and depressive symptomatology in psychotic disorders. Eur. Arch. Psychiatry Clin. Neurosci. https://doi.org/10.1007/s00406-018-0897-z (2018).
Ayesa-Arriola, R. et al. Low-density lipoprotein cholesterol and suicidal behaviour in a large sample of first-episode psychosis patients. World J. Biol. Psychiatry, 1–4, https://doi.org/10.1080/15622975.2017.1414305 (2018).
Costa, A. M. et al. Hormone profile in acute psychotic disorders: a cross-sectional comparison of serum hormone concentrations in treated and untreated male patients with schizophrenia. Curr. Ther. Res. Clin. Exp. 67, 350–363 (2006).
Dean, B., Digney, A., Sundram, S., Thomas, E. & Scarr, E. Plasma apolipoprotein E is decreased in schizophrenia spectrum and bipolar disorder. Psychiatry Res. 158, 75–78 (2008).
Stryer, L. Biochemistry 3rd edn, (W.H. Freeman, New York, 1988).
Acknowledgements
This study was supported by the Stanley Medical Research Institute (SMRI). We would like to thank the Department of Psychiatry, University of Cologne (Germany) and the Erasmus Medical Centre in Rotterdam (Netherlands) for provision of biological samples, in addition to support staff at the affiliated institutions. We would also like to thank Michael Lim.
Author information
Authors and Affiliations
Contributions
J.D.C. conceived, carried out the study, and wrote the first draft of the manuscript. S.Y.S.H. discussed the approach and critically contributed to the final version of manuscript. J.T. conducted the pathway analysis and participated in the first draft of the manuscript. S.O. and N.R. performed mass spectrometry and S.O. contributed to the final version of the manuscript. N.J.M.v.B. and F.M.L. collected clinical data and serum samples. S.B. supervised the study and critically contributed to the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
S.B. is a director of Psynova Neurotech Ltd and PsyOmics Ltd.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cooper, J.D., Han, S.Y.S., Tomasik, J. et al. Multimodel inference for biomarker development: an application to schizophrenia. Transl Psychiatry 9, 83 (2019). https://doi.org/10.1038/s41398-019-0419-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-019-0419-4
This article is cited by
-
Evaluating the Performance of Multi-criteria Decision-making Techniques in Flood Susceptibility Mapping
Journal of the Geological Society of India (2023)
-
A highly predictive autoantibody-based biomarker panel for prognosis in early-stage NSCLC with potential therapeutic implications
British Journal of Cancer (2022)
-
Moderators of reputation effects in peer-to-peer online markets: a meta-analytic model selection approach
Journal of Computational Social Science (2022)
-
Diagnostic model development for schizophrenia based on peripheral blood mononuclear cell subtype-specific expression of metabolic markers
Translational Psychiatry (2022)
-
A machine learning algorithm to differentiate bipolar disorder from major depressive disorder using an online mental health questionnaire and blood biomarker data
Translational Psychiatry (2021)
