
Abstract
One of the best performing semiconducting polymers used in bulk heterojunction devices is PCDTBT, a polycarbazole derivative with solar-conversion efficiencies as high as 7.2%. Here we report the formation of bilayer ordering in PCDTBT, and postulate that this structural motif is a direct consequence of the polymer's molecular design. This bilayer motif is composed of a pair of backbones arranged side-to-side where the alkyl tails are on the outer side. This is in stark contrast to the monolayer ordering found in other conjugated polymers. The crystalline bilayer phase forms at elevated temperatures and persists after cooling to room temperature. The existence of bilayer ordering, along with its high-packing fraction of conjugated moieties, may guide the synthesis of new materials with improved optoelectronic properties.
Similar content being viewed by others
Introduction
Conjugated polymers have been proposed for use in a wide variety of next generation technologies including displays, solid-state lighting, transistors and organic photovoltaic devices1,2,3, but relatively little is known about how these polymers pack with the exception of a few materials4. The active layer in organic photovoltaic bulk heterojunction devices is composed of a blend of π-conjugated, electron-donor polymers and electron-acceptor molecules, such as the fullerene derivative [6,6]-phenyl-C61-butyric acid methyl ester (PCBM). Whereas the optoelectronic properties of the electron-donor polymer are primarily determined by the choice of the conjugated backbone, the solubility is predominantly determined by the position, length and makeup of the alkyl side chains. Both the polymer backbone and the alkyl side chains influence device efficiency and molecular packing structure, especially in donor–acceptor copolymers, which contain an ordered sequence of different subunits5. Systematic studies of the effect of different side-chain groups, with the same backbone, have shown that the length and the type of alkyl chains affect the packing structure of the active layer, such as the π–π distance and polymer orientation, and these properties have been correlated with the device performances, fill factor, open circuit voltage and short circuit current. Such studies have been conducted for alternating silicon-bridged bithiophene and benzothiadiazole6, alternating thienothiophene and benzodithiophene (PTB)7, alternating naphtho[2,1-b:3,4-b']dithiophene and 4,7-di(thiophen-2-yl)benzothiadiazole (PNDT–DTBT)8, alternating bithiazole and benzodithiophene (BTz–BDT)9, and poly(3-alkylthiophene) (P3AT)10 among others. Decreasing the length (volume) of the alkyl chains has the potential to improve the polymer's charge mobility and optical density because these regions are resistive and do not contribute to optical absorption, but steric and other factors may lead to the opposite behaviour10.
The molecular structure of poly[N-9′′-hepta-decanyl-2,7-carbazole-alt-5,5-(4′,7′-di-2-thienyl-2′,1′,3′-benzothiadiazole)](PCDTBT), is shown in Fig. 1a. First synthesized by Leclerc and co-workers11, it is one of the most extensively studied donor–acceptor copolymers. The lone branched side chain along the ~2 nm backbone subunit-repeat distance is unusual as most polymers with such long repeat lengths have multiple alkyl chains with more alkyl chain material per polymer repeat unit. PCDTBT has been reported to have relatively high solubility11, nearly perfect internal quantum efficiency11, high-power conversion efficiency (>7.2%)12,13 in samples annealed below 80 °C and excellent thermal stability14, with operating lifetimes estimated to be ~7 years15. For thin films, no crystalline peaks were detected in the X-ray scattering profiles in studies (conventional source), for samples annealed as high as 400 °C (ref. 14). On this basis, an amorphous structure was reported. However, bulk PCDTBT showed the existence of two broad features, attributed to a layer spacing of 20.9 Å and a π–π stacking distance of 4.4 Å1616), much greater than the reported π–π distances of other successful polymers, such as 3.8 Å for Poly(3-hexylthiophene) (P3HT)17,18, and 3.55 Å for Poly[2,1,3-benzothiadiazole-4,7-diyl[4,4-bis(2-ethylhexyl)-4H-cyclopenta[2,1-b:3,4-b′]dithiophene-2,6-diyl]] (PCPDTBT)19. For many systems, a large π–π distance will hinder the electronic coupling and lead to poor device performance7. Despite the large π–π spacing and the reported amorphous-like structure, PCDTBT photovoltaic devices with thin active layers exhibit excellent performance. Additional structural features of PCDTBT, which may help provide a basis for the superior performance, have not been previously reported.

(a) The molecular structure of PCDTBT. (b) The experimental geometry of the grazing incidence wide-angle X-ray scattering geometry.
Results
X-ray scattering studies of PCDTBT films
Thin films, ~100 nm thick, made from PCDTBT were investigated, using Grazing incidence wide-angle X-ray scattering (GIWAXS) (Fig. 1b) under vacuum. To provide an overview of the scattering from the PCDTBT thin films, we show 4 PCDTBT scattering patterns in Fig. 2 at (a) 25 °C, (b) 140 °C, (c) 200 °C, and (d) after subsequent cooling to 25 °C. At 25 °C, before annealing (Fig. 2a), two weak and broad diffusive rings of scattering were observed along with a vertical streak that is associated with specular diffuse scattering. The PCDTBT scattering features, shown in Fig. 2a, were more defined than those reported previously under similar sample preparation conditions, using a much weaker, conventional X-ray source12. In both previous and present studies, the diffuse nature of the scattering indicates a rather disordered, amorphous-like state of the polymer film. For P3HT, a prototypical conjugated polymer, similar scattering rings, albeit somewhat better defined, have been associated with the distance between layers and to the π–π stacking distance1. By analogy, we also associate the ring at about 0.35 Å−1 in PCDTBT as originating from the layer spacing and the ring at 1.42 Å−1 with the π–π stacking distance. From Fig. 2a, the layering ring appears slightly elliptical-like rather than circular as it is located at ~0.36 Å−1 along the qz axis, and at ~0.33 Å−1 along the qr axis. These peak positions respectively correspond to layer spacings of ~17.5 and ~19.0 Å, indicating that the layer spacing is more compact when the layers stack along the qz (surface normal) direction, thus suggesting coexisting polymorphs. Coexisting polymorphs have also been observed in similar thin films materials such as PCPDTBT20. The observed PCDTBT layer spacings are smaller than the 20.9 Å spacing reported for bulk PCDTBT16. The π–π stacking ring (q~1.42 Å−1) corresponds to an average spacing of 4.4 Å, identical to that obtained for bulk samples16.

The images were obtained at (a) 25 °C before annealing; (b) 140 °C; (c) 200 °C; (d) 25 °C after annealing.
The scattering pattern at 140 °C (Fig. 2b) exhibits similar features as to the 25 °C pattern. However, the scattering ring associated with layering is both sharper and more intense than at 25 °C, suggesting enhanced order. The layering peak is somewhat aligned along the qz direction, thus suggesting a preferential 'edge-on' orientation. In addition, this scattering ring is now symmetric with a uniform radius of 0.302 Å−1, corresponding to a 20.8 Å layer spacing. This seems to suggest that the multiple polymorphs observed at lower temperatures have vanished. There is no apparent change in the peak positions and widths associated with the π–π stacking along the qz direction between 25 and 140 °C; however, the intensity gradually increases with increasing temperature. At 140 °C, above the 130 °C glass-transition temperature16, weak features start to emerge along the qz axis between the layering and the π–π peaks. These diffraction features correspond to the onset of a crystalline structure that is fully established by 200°C, as discussed below.
Well-defined Bragg peaks are observed at 200 °C (Fig. 2c) thus indicating the formation of a crystalline phase. We note that previous studies, albeit with a much weaker source (non-synchrotron), on PCDTBT films at similar temperatures failed to reveal the formation of a crystalline or oriented phase12. The diffraction peaks remain even after the film was cooled to 25 °C (Fig. 2d) and this provides strong evidence that the amorphous structure observed before annealing is a non-equilibrium structure. The layering peak at ~0.320 Å−1 along the qz axis, also indicating preferential 'edge-on' orientation, appears very similar to that observed at 140 °C, whereas the new spots at ~0.634 and ~0.947 Å−1, which emerge at around 160 °C (shown in Fig. 3), were not observed at 140 °C. The two new peaks are second- and third-order reflections; thus, the overall layering motif with a ~20 Å layer spacing does not change between 140 and 200°C. The appearance of the six off-axis peaks indicates the formation of long-range order structure parallel to the substrate and by 200 °C, they are located at (±0.150, 0.186), (±0.150, 0.494) and (±0.150, 0.811) Å−1. This long-range order must originate from crystallinity along the backbone direction as (1) the scattering associated with the π–π stacking is virtually unchanged between 140 and 200 °C and (2) the length scale 2π/0.150 Å−1≈41.9 Å is too long to be along the π–π stacking direction. The scattering pattern includes contributions from a crystalline phase with its well-defined Bragg peaks and a non-crystalline phase that gives rise to the more diffuse scattering features. These diffuse features do not affect the overall identification of the structure of the crystalline regions, the focus of the present investigation. Films prepared both by blade-coating and spin-casting, provided similar results and indicated no preferential backbone alignment with respect to the blade-coating direction.

The scattering profiles along qz obtained at qr~0 (lower seven curves) and qr=0.15 Å−1 (top two curves). The dashed lines (black) are fits to a Gaussian profile. The blue 25 °C curve is measured before thermal treatment. The orange 25 °C curve is measured after thermal treatment.
Unit cell calculation and the bilayer motif
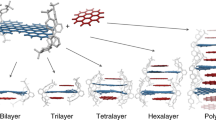
The diffraction pattern corresponding to the observed reflections is illustrated in Fig. 4a. We note that the pattern cannot be indexed by a rectangular or centred-rectangular unit cell, but rather by an oblique unit cell with b1=(0,0.333) Å−1 and b2=(0.150, 0.190) Å−1 (25 °C, after annealing). The real-space primitive unit cell vectors are a1=(−23.9, 18.9) and a2=(41.9, 0) Å (Fig. 4b), where the x axis is in-plane and the y axis is out-of-plane. The 41.9 Å length corresponds to the length of two linear backbone segments. Further, there is a constant shift of 23.9 Å between backbone chains separated by the 18.9 Å layer spacing. In Fig. 4c, we show the proposed molecular motif of the lamellar stacking structure of PCDTBT, which includes the pairing of neighbouring conjugated backbones to form bilayers (discussed below). In the 'molecular schematic', the alkyl chains are not explicitly drawn because there is no evidence based on the diffraction or Fourier transform infrared spectroscopy (FTIR) (Supplementary Fig. S1) of chain crystallinity, and because the chains are not confined to a single layer owing to the high density of gauche defects21,22. Figure 4c shows that the undulations between neighbouring bilayers are out-of-phase with each other and this allows the alkyl chains to aggregate giving the appearance of a chain of alkyl beads. As only short-range order is observed along the π–π stacking direction, there is no evidence for three-dimensional crystallization.

(a) Diffraction pattern obtained from the Bragg peaks shown in Fig. 2d. The filled circle corresponds to the origin. (b) The corresponding oblique real-space lattice obtained from the scattering pattern. The π–π direction is normal to this plane. (c) Undulating backbone configuration for one of possible conformations of a backbone obtained by ab initio calculations (Fig. 5c). The second chain is merely a rotated/translated copy of the first one. Including the full interactions would provide further adjustment in the positions perpendicular to the backbone. The alkyl chains (C8H17) are denoted by R for simplicity and occupy the blue-shaded region. This is the same oblique unit cell shown in (b). (d) A three-dimensional illustration of the crystalline phase where the red trapezoid identifies the 2D unit cell.
Packing and mass density considerations, as described below, clearly show that there must be four PCDTBT repeating units per unit cell, and this provides support for the pairing model shown in Fig. 4c. To determine the room temperature mass density, we calculated the volume of the unit cell to be 3,484 Å3, using the measured two-dimensional (2D) unit cell lattice parameters (18.9 and 41.9 Å) along with the average π–π stacking distance (4.4 Å). Placing two segments (MW=2×701) into the unit cell gives rise to a density of 0.67 g cm−3. If there are 4 PCDTBT segments per unit cell, the calculated density doubles to 1.34 g cm−3. Although direct density measurements were inaccurate because of bubble formation, the density is expected to be higher than the mass density of P3HT (1.10 g cm−3)17. A centred arrangement formed by including an additional polymer backbone between the layers is inconsistent with the diffraction pattern, as this centred lattice would give rise to missing diffraction peaks that are observed. This indicates that the layers are composed of conjugated backbone pairs, rather than a single backbone as with P3HT, thus supporting the bilayer arrangement shown in Fig. 4c,d.
In the following, we introduce a simple method of calculating the absolute volume contributions from the alkyl and the conjugated regions into the unit cell of conjugated polymers and, as described below, this approach supports the monolayer designation for P3HT and the bilayer designation for PCDTBT. For each polymer repeat segment, we assign atoms in the two octyl chains to the alkyl region and the remaining atoms to the conjugated region. The thickness of the alkyl region is calculated from the ratio of the alkyl volume (calculated from its bulk n-alkane values) to the cross-sectional area of the unit cell transverse to the layering direction, where we have assumed that the alkyl chains are space filling. For PCDTBT, this transverse cross-sectional area is 41.9×4.4 Å and each octyl chain has a volume of 240 Å3, half the volume of a liquid n-hexadecane molecule. If we assume a monolayer motif (two PCDTBT segments per unit cell), an alkyl region layer thickness of 5.2 Å is obtained. A conjugated region layer, with a thickness of 13.7 Å, is obtained from the difference between the 18.9 Å layer spacing and the 5.2 Å alkyl region layer thickness. This is more than 3 times the 4.1 Å P3HT conjugated thickness obtained using a similar calculation taking into account a layer spacing of 16.8 Å and a π–π stacking distance of 3.8 Å for P3HT (ref. 17). For the proposed PCDTBT bilayer model (4 PCDTBT segments per unit cell) shown in Fig. 4c, the calculated alkyl region layer thickness is 2×5.2=10.4 Å, thus leaving a conjugated thickness of 8.5 Å for a pair of backbone segments, a reasonable thickness compared with the unphysical, 13.8 Å thickness calculated for the monolayer model above. This conjugated region thickness is about twice the calculated 4.1 Å P3HT thickness (see above). Finally, for the proposed pairing model shown in Fig. 4c,d there is a sinusoidal-like variation in the position of the backbone along the layering direction and this feature explains the absence of higher order diffraction peaks along qr.
First principles calculation of backbone conformations
The precise details of the pairing model can not be ascertained from the X-ray data alone, because there are too few reflections to provide a crystallographic analysis. Additional structural insight on the backbone configuration was obtained from the ab initio calculations described below. All calculations were performed using the GAUSSIAN 09 package23. The dimer (two backbone repeat units) of PCDTBT with a methyl side chain (Fig. 5A) was first optimized using the B3LYP functional and the 6-311g(d) basis set. The potential energy surfaces of the two dihedrals (benzothiadiazole(BTD)-thiophene(THI) and carbazole(CAR)-THI) were then scanned using the same functional and basis set. The results, shown in Fig. 5B, indicate that the BTD and the THI units prefer to have dihedral angle of φ=0, though the second minimum of φ=π is only 2.9 kJ per mol (1.15 kBT at room temperature) higher. The π-systems of the CAR and the THI repeat units are not parallel to each other, also the two minima at ~30 and 150° have practically the same energy. One can conclude that there is practically no preference for the backbone to have trans- or cis- conformations between the repeat units, and self-assembly is mostly driven by the competition between packing of side chains and van der Waals interactions between backbones.

(A) Scanned dihedrals: the shown BTD–THI (red) dihedral angle is φ=0 and CAR–THI (blue) angle is φ=π. (B) Potential energy surfaces of scanned dihedrals. (C) Planar molecular conformations optimized in periodic boundary conditions.
The backbone conformation within the proposed unit cell was also calculated. The dimer (Fig. 5A) has seven dihedral angles and hence 27 distinct conformers. As a 180° flip of the BTD group does not practically change the polymer conformation, this number is further reduced to 25 conformers. Most of the conformations are either incompatible with the periodic boundary conditions, or have to significantly twist out of the plane to accommodate them, hence only a few configurations were suitable.
To select the suitable conformers, all distinct (32) molecular conformations were optimized in one-dimensional periodic boundary conditions using the B3LYP functional and the 6-311g(d) basis set. Four practically planar conformations with alkyl side chains on one side of the backbone are shown in Fig. 5C. Out of these four (labelled a,b,c,d in the Fig. 5C), one can construct the proposed unit cell using the period-matching pairs, such as a–a, a–b, b–b, c–c, c–d, or d–d. For the a–a, b–b and c–c pairs, one of the chains has to be flipped to avoid the overlap of side chains. One of these possible configurations is shown in the Fig. 4c.
Temperature dependence of PCDTBT thin films
To investigate the temperature evolution behaviour in detail, we have examined and analysed the scattering profiles along the qz axis and these were extracted from the 2D scattering patterns such as those shown in Fig. 2 (acquired every 20 °C). Representative scattering profiles, displayed as the lowest seven curves in Fig. 3 after subtraction of the diffuse signal, were obtained slightly off-axis to minimize the specular diffuse background. The Gaussian model fits, dashed lines displayed in Fig. 3, provide an excellent representation of the scattering profiles. The resulting fitting parameters are summarized in Table 1, where the layer spacing (a0) was calculated from the Gaussian's centre (q0) by a0=2π/q0 and the coherence length (ξ), a value associated with the average grain size, was calculated from the standard deviation of the Gaussian fits (σ) by ξ=π/σ. Over the temperature range from 25 to 100°C, the layering peak is very broad and the analysis gives nearly temperature-independent coherence lengths and layer spacings, ~30±3 and ~17.1±0.4 Å, respectively. Here the coherence length is only a few layers thick, similar to that of a liquid, and supports the designation that the pre-annealed sample is amorphous14. Despite an increase in the scattering intensity with increasing temperature below 100 °C, there is no change in the overall structural motif. However, at 140 °C, above the glass-transition temperature, the layering peak position is shifted to 0.302 Å−1. This large shift in the layer spacing, ~3 Å, is too large to be attributed to a simple lattice expansion, but rather it corresponds to a phase change. Further, this peak has narrowed corresponding to a coherence length of 78 Å. The second-order peak appears at 160°C and the third-order peak appears at 200 °C. By 200 °C, the scattering profiles have sharpened further and the coherence length reaches 123 Å corresponding to about 6 layers, still about a factor of two less correlated than for P3HT at the same temperature24. The first order peak shifts slightly to higher qz when the temperature is raised from 140 to 200 °C, corresponding to a lattice contraction. This contraction may result from the ability of the layers to pack more efficiently in the crystalline phase. On cooling to 25 °C, the scattering patterns, Fig. 2d, remained the same as at 200 °C despite the 3.6% lattice contraction, exemplified by the shift in the peak positions, Fig. 3 (orange and yellow). This is much less than the 7% contraction observed for P3HT over this same temperature range25. We attribute this larger P3HT coefficient to the higher volume fraction of alkyl chains in P3HT.
We have also examined the thermal behaviour of the off-axis peaks. On cooling to 25 °C, we observe no significant change in the qr positions (0.150 Å−1) of the peaks. Thus, the scattering profiles of the off-axis peaks along qz are obtained from the 2D scattering images at qr=0.150 Å−1, shown by the top two curves in Fig. 3. The thermal behaviours of the off-axis peaks are very similar to the on-axis peaks. Together, our results show that the thermal expansion is along the layering direction and not along the backbone direction.
X-ray scattering studies of PCDTBT/PCBM blends
Figure 6 shows GIWAXS images of thin films of PCDTBT blended with PCBM at (a) 25 °C before annealing, (b) 25 °C after annealing and (c) the scattering profiles along qz at different temperatures. The 1:4 ratio of PCDTBT to PCBM is identical to that used in high-efficiency bulk heterojunction devices, reported previously12,13. Because of the high concentration of PCBM, the amorphous PCBM ring at around 1.4 Å−1 dominates the scattering image, whereas the weak layering and π–π stacking peaks of the polymer are barely visible at 25 °C, before annealing (Fig. 6a).

The GIWAXS images are obtained at (a) 25 °C, before annealing, and (b) 25 °C, after annealing. (c) The corresponding scattering profiles along qz. The inset shows the region of interest near 0.36 Å−1, at temperatures at 140 °C and below. The blue and orange 25 °C curves are measured before and after thermal treatment, respectively.
After annealing, shown in Fig. 6b, the sharp peaks observed are from the crystallization of PCBM, consistent with previous results26. The scattering profiles along qz (Fig. 6c) reveal details of the temperature-dependent structural evolution. Below 140 °C, the lowest order-layering peak is very weak (see inset) but it is located at ~0.36 Å−1, the same position as in the pure PCDTBT bilayer phase. This suggests the appearance of the bilayer phase in the blend material below 140 °C. The other crystalline peaks that indicate long-range order are not observed in the blends. At temperatures above 160 °C, a pronounced layering peak emerges in the blend corresponding to a ~15.7 Å layer spacing, too small for the bilayer phase. At these elevated temperatures, the rings of scattering from the PCBM are no longer isotropic after annealing suggesting orientational order that maybe induced by the PCDTBT crystallinity. This might correspond to a mixed PCDTBT/PCBM phase, such as observed for PBTTT blends27. Thus, our results suggest that a disordered phase with a local bilayer motif may account for the excellent cell performance observed in the blends below 140 °C.
Discussion
To our knowledge, our proposed backbone pairing mode, composed of side-to-side backbones has not been previously reported for conjugated polymers. The physical motivation for the formation of the bilayer polymer backbone model can be found in block copolymers, polymers with a regular series of two different blocks. If the two blocks are relatively immiscible with each other, then the system undergoes a nanophase separation between the two blocks. Depending on the strength of the repulsive interactions, this may lead to lamellar, spherical or hexagonally packed phases28. Phospholipids also form a bilayer motif to nanophase separate their ionic (hydrophilic) and alkyl (hydrophobic) regions. For conjugated polymers, the alkyl regions would also like to phase separate from the backbone and for polymers such as regioregular P3HT; this leads to monolayer lamellar ordering, in part, because the alkyl chains must alternate from either side due to steric constraints. As, for PCDTBT, the alkyl chains are all anchored on the same segment, this allows the alkyl-free side of the PCDTBT backbones to pair. This arrangement is favoured, because it minimizes the interfacial energy between the nonpolar alkyl side chains and the polar backbones.
Developing structure-function relationships, in conjugated polymers, is essential for improving device performance in organic photovoltaic devices29. Our results show that one of the best performing device materials, PCDTBT, exhibits a bilayer structural motif described by conjugated backbone pairs with the alkyl chains filling the interstitial regions between the pairs. The bilayer motif may result from the lone attachment site of the alkyl chains on the backbone and it seems to be an equilibrium property of PCDTBT, because it is found in films at high temperature and at room temperature after annealing. The broad diffraction peaks observed in films before annealing, both in the pure material and in the blends, indicates amorphous-like behaviour and suggests a random network of bilayer pairs. Under these conditions, excellent device performance is observed. A better understanding of the bilayer motif in conjugated polymers and its relationship to device performance, especially in other materials, may aid in the design of new high-performance polymers for the next generation of transistor, light-emitting diode, and solar-cell applications.
Methods
Preparation of PCDTBT thin films
The PCDTBT, supplied by Konarka, has Mw~100 k, a polydispersity index of 2.75, and a glass-transition temperature of 130 °C, consistent with previous measurements16. The thin films were prepared by blade-coating 8 mg ml−1 PCDTBT in 1,2-dichlorobenzene (oDCB) at 80 °C on silicon wafers.
X-ray scattering measurements
The GIWAXS experiments were performed at the X9 undulator beamline at the National Synchrotron Light Source, Brookhaven National Laboratory, where the 14.0 keV photons (λ=0.0886 nm) are focused to a spot with a height of 80 μm and a width of 200 μm, at the sample position. The X-ray incident angle was set to ~0.15°, an angle above the critical angle of the polymer so as to assure a full penetration into the film. The diffraction images, such as those shown in Fig. 2, reveal the sample's out-of-plane structure (normal to the substrate) along vertical axis (qz) and in-plane structure (parallel to the substrate) along horizontal axis (qr). The scattering/sample chamber vacuum was maintained at ~10−2 torr at all temperatures to reduce thermal sample degradation, beam damage, diffuse scattering and X-ray absorption. We have accounted for the intersection of Ewald sphere and reciprocal space in the extracted scattering profiles and the measured peak positions.
FTIR spectra
The aliphatic region of the FTIR spectra for PCDTBT, both as-cast and annealed, is shown in Supplementary Figure S1. PCDTBT was cast on undoped, zone-cast, double-side polished silicon wafers with resistivity >6,000 ohms cm−1, <100>orientation, 380–410 μm thickness, (WRS Materials). Spectra were collected in transmission mode at Brewster's angle (Θ=16°) with a DTGS detector, using a Nicolet model 6,700 FTIR spectrometer (ThermoFisher) equipped with a KRS-5 polarizer (Spectra-Tech). Each spectrum represents the sum of 256 individual spectra, which were subjected to background correction, relative to a representative blank silicon chip. The corrected spectra were then fit to a second-order baseline and the peak locations and widths were modelled with a combination of Lorentzians using spectral analysis macros written by B. and P.U.P.A. Gilbert for the software package IGOR Pro 6 (Wavemetrics)30. To determine the exact peak positions of each vibrational mode, we modelled the observed spectra with a combination of six Lorentzians21. Most relevant to this work was the peak position of the methylene anti-symmetric stretch at 2,928 cm−1. For extended alkyl chains, this vibrational mode is known to be sensitive to the degree of gauche defects present in the alkyl chain22. The position of this peak in the as-cast spectra is consistent with a 'liquid-like' alkyl side-chain (that is, one with a significant number of gauche defects). On thermal annealing, this peak position does not change, suggesting that the side-chains of PCDTBT exist in a disordered state, independent of the crystallinity of the backbones.
Additional information
How to cite this article: Lu, X. et al. Bilayer order in a polycarbazole-conjugated polymer. Nat. Commun. 3:795 doi: 10.1038/ncomms1790 (2012).
References
Sirringhaus, H. et al. Two-dimensional charge transport in self-organized, high-mobility conjugated polymers. Nature 401, 685–688 (1999).
McCulloch, I. et al. Liquid-crystalline semiconducting polymers with high charge-carrier mobility. Nature Mater. 5, 328–333 (2006).
Yan, H. et al. A high-mobility electron-transporting polymer for printed transistors. Nature 457, 679–686 (2009).
Winokur, M. J. & Chunwachirasiri, W. Nanoscale structure-property relationships in conjugated polymers: Implications for present and future device applications. J. Polym. Sci B Polym. Phys. 41, 2630–2648 (2003).
Heeger, A. J. Semiconducting polymers: the third generation. Chem. Soc. Rev. 39, 2354–2371 (2010).
Coffin, R. C., Peet, J., Rogers, J. & Bazan, G. C. Streamlined microwave-assisted preparation of narrow-bandgap conjugated polymers for high-performance bulk heterojunction solar cells. Nat. Chem. 1, 657–661 (2009).
Szarko, J. M. et al. When function follows form: Effects of donor copolymer side chains on film morphology and BHJ solar cell performance. Adv. Mater. 22, 5468–5472 (2010).
Yang, L., Zhou, H. & You, W. Quantitatively analyzing the influence of side chains on photovoltaic properties of polymer-fullerene solar cells. J. Phys. Chem. C 114, 16793–16800 (2010).
Shi, Q. et al. Side chain engineering of copolymers based on bithiazole and benzodithiophene for enhanced photovoltaic performance. Macromolecules 44, 4230–4240 (2011).
Gadisa, A. et al. Effect of alkyl side-chain length on photovoltaic properties of poly(3-alkylthiophene)/PCBM bulk heterojunctions. Adv. Funct. Mater. 19, 3300–3306 (2009).
Blouin, N., Michaud, A. & Leclerc, M. A low-bandgap poly(2,7-carbazole) derivative for use in high-performance solar cells. Adv. Mater. 19, 2295–2300 (2007).
Park, S. H. et al. Bulk heterojunction solar cells with internal quantum efficiency approaching 100%. Nat. Photon 3, 297–302 (2009).
Sun, Y. et al. Efficient, air-stable bulk heterojunction polymer solar cells using moox as the anode interfacial layer. Adv. Mater. 23, 2226–2230 (2011).
Cho, S. et al. A thermally stable semiconducting polymer. Adv. Mater. 22, 1253–1257 (2010).
Peters, C. H. et al. High efficiency polymer solar cells with long operating lifetimes. Adv. Energy Mater. 1, 491–494 (2011).
Blouin, N. et al. Toward a rational design of poly(2,7-carbazole) derivatives for solar cells. J. Am. Chem. Soc. 130, 732–742 (2008).
Prosa, T. J., Winokur, M. J., Moulton, J., Smith, P. & Heeger, A. J. X-ray structural studies of poly(3-alkylthiophenes): an example of an inverse comb. Macromolecules 25, 4364–4372 (1992).
Chang, J.- F. et al. Enhanced mobility of poly(3-hexylthiophene) transistors by spin-coating from high-boiling-point solvents. Chem. Mater. 16, 4772–4776 (2004).
Park, Y. D. et al. Solubility-controlled structural ordering of narrow bandgap conjugated polymers. Adv. Energy Mater. 1, 63–67 (2011).
Rogers, J. T., Schmidt, K., Toney, M. F., Kramer, E. J. & Bazan, G. C. Structural order in bulk heterojunction films prepared with solvent additives. Adv. Mater. 23, 2284–2288 (2011).
Gurau, M. C. et al. Measuring molecular order in poly(3-alkylthiophene) thin films with polarizing spectroscopies. Langmuir 23, 834–842 (2007).
Snyder, R. G., Strauss, H. L. & Elliger, C. A. Carbon-hydrogen stretching modes and the structure of n-alkyl chains 1. long, disordered chains. J. Phys. Chem. 86, 5145–5150 (1982).
Frisch, M. J. et al. Gaussian 09 Revision A. 1 (Gaussian Inc., Wallingford CT, 2009).
Hlaing, H. et al. Nanoimprint-induced molecular orientation in semiconducting polymer nanostructures. ACS Nano 5, 7532–7538 (2011).
Lilliu, S. et al. Dynamics of crystallization and disorder during annealing of P3HT/PCBM bulk heterojunctions. Macromolecules 44, 2725–2734 (2011).
Yang, X. et al. Crystalline organization of a methanofullerene as used for plastic solar-cell applications. Adv. Mater. 16, 802–806 (2004).
Mayer, A. C. et al. Bimolecular crystals of fullerenes in conjugated polymers and the implications of molecular mixing for solar cells. Adv. Funct. Mater. 19, 1173–1179 (2009).
Bates, F. & Fredrickson, G. Block copolymer thermodynamics: Theory and experiment. Annu. Rev. Phys. Chem. 41, 525–557 (1990).
Brabec, C. J., Heeney, M., McCulloch, I. & Nelson, J. Influence of blend microstructure on bulk heterojunction organic photovoltaic performance. Chem. Soc. Rev. 40, 1185–1199 (2011).
P.U.P.A. Gilbert 'GG Macros', http://home.physics.wisc.edu/gilbert/ (2012).
Acknowledgements
This research is supported by the US. Department of Energy, Basic Energy Sciences, by the Materials Sciences and Engineering Division (X.L, H.H. D.G. & B.O.) which is supported under Contract No. DE-AC02-98CH10886 and by Konarka Technologies. This work was partially supported by Energy Laboratory Research and Development Initiative at Brookhaven National Laboratories. This work was also supported by the DFG program IRTG 1404, DFG grant SPP 1355, and BMBF grant MESOMERIE. The authors wish to thank Mario Leclerc, Gilles Dennler, Barney Grubbs, Kevin Yager and Chuck Black for valuable discussions.
Author information
Authors and Affiliations
Contributions
X.L., H.H. and B.M.O. carried out the x-ray measurements. X.L., B.M.O. and W.H.J. developed the bilayer model. J.P. was responsible for preparing the samples. D.S.G. carried out the FTIR measurements. D.A. and K.K. carried out the ab initio calculations. All authors contributed to the editing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Description: Supplementary Figures S1 (PDF 69 kb)
Rights and permissions
About this article
Cite this article
Lu, X., Hlaing, H., Germack, D. et al. Bilayer order in a polycarbazole-conjugated polymer. Nat Commun 3, 795 (2012). https://doi.org/10.1038/ncomms1790
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms1790
This article is cited by
-
Argo: a data analysis program for quantum chemical calculations
Journal of Molecular Modeling (2019)
-
Rhodanine flanked indacenodithiophene as non-fullerene acceptor for efficient polymer solar cells
Science China Chemistry (2017)
-
Absorption-induced scattering and surface plasmon out-coupling from absorber-coated plasmonic metasurfaces
Nature Communications (2015)
-
High-performance ternary blend polymer solar cells involving both energy transfer and hole relay processes
Nature Communications (2015)
-
Efficient Organic Photovoltaics Utilizing Nanoscale Heterojunctions in Sequentially Deposited Polymer/fullerene Bilayer
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
