
Abstract
Activation of oncogenes by mechanisms other than genetic aberrations such as mutations, translocations, or amplifications is largely undefined. Here we report a novel isoform of the anaplastic lymphoma kinase (ALK) that is expressed in ∼11% of melanomas and sporadically in other human cancer types, but not in normal tissues. The novel ALK transcript initiates from a de novo alternative transcription initiation (ATI) site in ALK intron 19, and was termed ALKATI. In ALKATI-expressing tumours, the ATI site is enriched for H3K4me3 and RNA polymerase II, chromatin marks characteristic of active transcription initiation sites1. ALKATI is expressed from both ALK alleles, and no recurrent genetic aberrations are found at the ALK locus, indicating that the transcriptional activation is independent of genetic aberrations at the ALK locus. The ALKATI transcript encodes three proteins with molecular weights of 61.1, 60.8 and 58.7 kilodaltons, consisting primarily of the intracellular tyrosine kinase domain. ALKATI stimulates multiple oncogenic signalling pathways, drives growth-factor-independent cell proliferation in vitro, and promotes tumorigenesis in vivo in mouse models. ALK inhibitors can suppress the kinase activity of ALKATI, suggesting that patients with ALKATI-expressing tumours may benefit from ALK inhibitors. Our findings suggest a novel mechanism of oncogene activation in cancer through de novo alternative transcription initiation.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Accession codes
Primary accessions
European Nucleotide Archive
Sequence Read Archive
Data deposits
The sequence of ALKATI has been deposited in the European Nucleotide Archive under the accession number LN864494. RNA-seq, ChIP-seq, DNA and bisulfite sequencing data have been deposited in the NCBI Sequence Read Archive with the accession number SRP058714.
References
Kouzarides, T. Chromatin modifications and their function. Cell 128, 693–705 (2007)
Anders, S., Reyes, A. & Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017 (2012)
GTEx Consortium The Genotype-Tissue Expression (GTEx) project. Nature Genet. 45, 580–585 (2013)
Reis, P. P. et al. mRNA transcript quantification in archival samples using multiplexed, color-coded probes. BMC Biotechnol. 11, 46 (2011)
Northcott, P. A. et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511, 428–434 (2014)
Xie, M. et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nature Genet. 45, 836–841 (2013)
Calo, E. & Wysocka, J. Modification of enhancer chromatin: what, how, and why? Mol. Cell 49, 825–837 (2013)
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011)
Soda, M. et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566 (2007)
Lemmon, M. A. & Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 (2010)
Bresler, S. C. et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci. Transl. Med. 3, 108ra114 (2011)
Mossé, Y. P. et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455, 930–935 (2008)
Janoueix-Lerosey, I. et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455, 967–970 (2008)
Passoni, L. et al. Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Cancer Res. 69, 7338–7346 (2009)
Montavon, G. et al. Wild-type ALK and activating ALK-R1275Q and ALK-F1174L mutations upregulate Myc and initiate tumor formation in murine neural crest progenitor cells. Oncotarget 5, 4452–4466 (2014)
Schulte, J. H. et al. High ALK receptor tyrosine kinase expression supersedes ALK mutation as a determining factor of an unfavorable phenotype in primary neuroblastoma. Clin. Cancer Res. 17, 5082–5092 (2011)
Cheng, D. T. et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 17, 251–264 (2015)
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009)
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009)
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010)
Robinson, J. T. et al. Integrative genomics viewer. Nature Biotechnol. 29, 24–26 (2011)
Carbon, S. et al. AmiGO: online access to ontology and annotation data. Bioinformatics 25, 288–289 (2009)
Mathelier, A. et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42, D142–D147 (2014)
Chi, P. et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature 467, 849–853 (2010)
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008)
Won, H. H., Scott, S. N., Brannon, A. R., Shah, R. H. & Berger, M. F. Detecting somatic genetic alterations in tumor specimens by exon capture and massively parallel sequencing. J. Vis. Exp. e50710 (2013)
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009)
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genet. 43, 491–498 (2011)
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnol. 31, 213–219 (2013)
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012)
Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011)
Boeva, V. et al. Control-free calling of copy number alterations in deep-sequencing data using GC-content normalization. Bioinformatics 27, 268–269 (2011)
Wang, J. et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nature Methods 8, 652–654 (2011)
McLaren, W. et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070 (2010)
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009)
Geiss, G. K. et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nature Biotechnol. 26, 317–325 (2008)
Bennett, D. C., Cooper, P. J. & Hart, I. R. A line of non-tumorigenic mouse melanocytes, syngeneic with the B16 melanoma and requiring a tumour promoter for growth. Int. J. Cancer 39, 414–418 (1987)
Refaeli, Y., Van Parijs, L., Alexander, S. I. & Abbas, A. K. Interferon gamma is required for activation-induced death of T lymphocytes. J. Exp. Med. 196, 999–1005 (2002)
Ponomarev, V. et al. A novel triple-modality reporter gene for whole-body fluorescent, bioluminescent, and nuclear noninvasive imaging. Eur. J. Nucl. Med. Mol. Imaging 31, 740–751 (2004)
Acknowledgements
We thank P. Romanienko for the northern blot; W. Pao for the EML4–ALK construct; L. Garraway for the 501Mel and WW94 cell lines; P. Koppikar for the Ba/F3 cells; and C. Sawyers, O. Abdel-Wahab, K. Griewank, and O. Guryanova for reviewing the manuscript. Next generation sequencing, array CGH and NanoString nCounter Assay were performed at the Center for Molecular Oncology, and interphase fluorescence in situ hybridization in the Molecular Cytogenetics Core at Memorial Sloan Kettering Cancer Center. This work was supported by grants from the Harry J. Lloyd Trust, the Jubiläumsfonds of the Oesterreichische Nationalbank (15461) and a Charles H. Revson Senior Fellowship to T.W.; the National Institutes of Health (NIH) grants DP2CA174499 and K08CA151660, and the Geoffrey Beene Cancer Research Fund to P.C.; the NIH grant K08CA140946, Alfred Bressler Scholars Endowment Fund and Gerstner Young Investigator Award to Y.C.; the NIH grant P50CA172012 to J. A. F.; the NIH grant P01CA12943 to M.L., M.F.B, L.W. and L.B.; and the NIH grant P30 CA008748 to Memorial Sloan Kettering Cancer Center (Core Grant).
Author information
Authors and Affiliations
Contributions
Experimental design: T.W., P.C. and Y.C. Sample collection: I.L., K.J.B., M.L., T.H., J.A.F., G.K.S., L.W., T.M. and R.M. 5′-RACE, array CGH, FISH and immunohistochemistry: T.W. Preparation of DNA and cDNA libraries and bisulfite sequencing: T.W. and S.N.S. Data analyses: T.W., W.L., M.B., R.S., A.S., N.L. and D.Z. NanoString: T.W. and L.B. Immunofluorescence: T.W. Western blots, and immunoprecipitation: T.W., Q.F.Z. and J.B. ChIP and ChIP-seq: L.R. and S.S. Generation of the expression vectors: T.W., Q.F.Z. and Z.C. Luciferase reporter assay: E.W.P.W. In vivo assays: T.W., A.C.O. and D.A.M. FACS: T.W. and W.H. In vitro kinase assay: T.W. and D.G. Review of histology and immunohistochemistry: T.W., K.B. and R.M. Patient data: M.A.P. and C.E.A. Manuscript writing: T.W., Y.C. and P.C. All authors reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
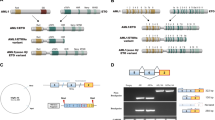
Extended Data Figure 1 Comparison of the RNA-seq profiles of various ALK transcripts.
RNA-seq data are displayed in the Integrative Genomics Viewer (IGV). The grey bars/arrows indicate the sequencing reads. The blue lines connect sequencing reads that are aligned over the splice site of joining exons. a, The ALKATI transcript shows expression of ALK exons 20–29 and of ∼400 bp in intron 19 (blue shaded area). No expression of exon 1–19 or intronic areas, other than in intron 19, are observed. The detailed view illustrates that the sequencing reads align continuously between exon 20 and intron 19 indicating uninterrupted transcription. The 5′-UTR of ALKATI (intron 19) and exon 20–29 are expressed at comparable levels. b, The full-length wild-type ALK transcript shows expression of all ALK exons and only very little expression of the introns. The detailed view displays that the sequencing reads align sharply to the exons, but not to the intron 19 region, which is present in ALKATI (blue shaded area). c, The ALK fusion transcript of a non-small cell lung cancer with an EML4–ALK translocation shows expression of ALK exons 20–29, and little expression of exons 1–19 and all introns. The detailed view illustrates that the transcription starts mainly at exon 20 due to a preserved splice site. Only few reads are aligned to the intron 19 region (blue shaded area). The green-labelled reads highlight chimaeric read pairs indicating the EML4–ALK translocation.
Extended Data Figure 2 Identification of the ALKATI transcript.
a, Northern blot of wild-type ALK-expressing neuroblastoma cell lines (SK-N-DZ and SK-N-BE2), EML4–ALK-expressing lung cancer cell lines (H3122, variant (V) 1 and H2228, variant (V) 3), ALKATI-expressing melanoma, one anaplastic thyroid carcinoma (ATC-28), and negative controls (melanoma cell lines). Except for the negative controls, each lane shows two bands: the lower B-band matches the shorter canonical (RefSeq) ALK transcript ending at ∼chr2:29,415,640; the upper A-band corresponds to a transcript with a 1.8 kb longer 3′-UTR ending at ∼chr2:29,413,840. Two ALKATI-expressing melanomas, MM-284 and MM-74, show only weak signals because less than 1 μg RNA was available; for all other samples 5–10 μg RNA were used. See Supplementary Fig. 1 for uncropped blots. b, RNA-seq data displayed in IGV. The Sashimi plot illustrates the shorter B and the longer A ALK transcripts by the sharp drop of sequencing reads in the 3′-UTR at chr2:29,415,640 for the B and at chr2:29,413,840 for the A transcript. c, IGV view of the 5′-RACE-cDNA fragments obtained by massively parallel sequencing. More than 95% of the sequencing reads (grey arrows) start within the main ATI site of 25 bp (hg19 chr2:29,446,744–29,446,768). d, Sanger sequencing of the cloned 5′-RACE-cDNA fragments confirms the continuous transcription starting in ALK intron 19 and extending to exons 20 and 21. e, The ALKATI transcript consists of ∼400 bp upstream of exon 20 and of ALK exons 20–29. The transcriptional initiation site was defined as the first base pair at which more than 5% of the transcripts were initiated (chr2:29,446,766). Other major transcription initiation sites are marked in red, the 5′- and 3′-UTRs in dark blue, the coding DNA sequence (CDS) in black, and the first and last base of each exon in light blue. The translation is initiated at 3 start codons (ATGs; bold and underlined): first ATG, hg19 chr2:29,446,360–29,446,362; second ATG, (+ 7–9); and third ATG (+ 61–3). f, The amino acid sequence of ALKATI. The translation is initiated at 1 of 3 start codons. The corresponding 3 methionines (bold and underlined) result in 3 different proteins, 61.08 kDa (552 amino acids), 60.82 kDa (550 amino acids), and 58.71 kDa (532 amino acids). The kinase domain is highlighted in red. The lysine in the ATP binding domain is marked bold and underlined, and was mutated to methionine (referring to wild-type ALK: p.K1150M) in the kinase-dead ALKATI-KD.
Extended Data Figure 3 RNApol II and H3K4me3 are enriched at the ATI site of ALKATI-expressing tumour samples.
a, b, ChIP–qPCR of H3K4me3 (a) and RNApol II (b) at the ATI site demonstrating enrichment of both marks in the ALKATI-expressing human tumour samples, but not in the negative controls, including a lung cancer cell line with EML4–ALK translocation (H3122) and a melanoma cell line (SKMEL-524). Error bars show mean ± s.e.m.; n = 3 technical replicates.
Extended Data Figure 4 ALKATI is transcribed from a genomically intact ALK locus.
a, Interphase FISH with ALK flanking probes demonstrates juxtaposed green and orange signals indicating no ALK rearrangement in MM-15. Scale bar, 10 µm. b, Interphase FISH in MM-74 shows 3 green/orange fusion signals in the majority of nuclei indicating a trisomy 2, but no ALK rearrangement. Scale bar, 10 µm. c, The top panel shows the genome-wide array CGH profile of MM-15 with numerous chromosomal gains and losses across the entire genome. The chromosomes are aligned along the x axis. The blue line illustrates the relative copy number (log2 ratio) and the blue bars highlight copy number gains and losses. The middle panel illustrates the relative copy number (blue line) of chromosome 2. Distal to the ALK locus, a loss on the short (p) arm of chromosome 2 is indicated. The lower panel illustrates the relative copy number across the ALK locus. The red and green squares represent the log2 ratio of individual array CGH probes (green, positive log2 ratio; red, negative log2 ratio). No disruption or selective gains or losses are found at the ALK locus. d, The genome-wide array CGH profile of MM-74 shows numerous chromosomal gains and losses across the entire genome in the top panel. The middle panel displays a relative copy number gain of the entire chromosome 2, which is in line with the trisomy of chromosome 2 as indicated by FISH. The lower panel also displays trisomy of chromosome 2, but indicates no focal gains and losses at the ALK locus.
Extended Data Figure 5 Targeted sequencing and whole-genome sequencing reveals no recurrent genomic aberrations at the ALK locus.
a, Ultra-deep sequencing data of the ALK locus are displayed in IGV. The genomic region around intron 19 reveals several single nucleotide variations (SNVs). However, the vast majority of SNVs at the ALK locus are also found in the general population as they are detected in the pool of normal DNA, which was used as the control (pooled normal, bottom panel). Numerous SNVs are also documented in the Single Nucleotide Polymorphism database (dbSNP; http://www.ncbi.nlm.nih.gov/SNP/). No genomic aberrations were found at the transcription initiation site of ALKATI. Supplementary Table 2 shows the detected SNVs and indels at the ALK locus after filtering out SNPs documented in the dbSNP database. None of the genomic aberrations was found in more than one case, indicating that the expression of ALKATI is probably not caused by alterations of the DNA nucleotide sequence. b, c, Circos plots of the whole-genome sequencing data of MM-15 (b) and ATC-28 (c) illustrating numerous SNV and structural aberrations. Supplementary Table 3 lists the detected single nucleotide polymorphisms, and Supplementary Table 4 the detected structural aberrations. No recurrent genomic aberrations were found at the ALK locus.
Extended Data Figure 6 Local chromatin context at the alternative transcription initiation (ATI) site.
a, UCSC Genome Browser view of the ATI site. The RepeatMasker track shows transposable elements at the ATI region, including a long-terminal repeat (LTR) in intron 19 (LTR16B2) and a long interspersed element (LINE) in intron 18. The ENCODE tracks reveal a DNase I hypersensitivity cluster and H3K4me1 enrichment, but no H3K27ac enrichment. b, The methylation status of the ALK locus was assessed by custom capture of the ALK locus, followed by bisulfite treatment and next-generation sequencing. Bisulfite sequencing results of H3122 (top) and MM-15 (bottom) are displayed in the CG-bisulfite mode of IGV. The red colour denotes ‘C’ (cytosine) corresponding to methylated cytosine, which is preserved during the bisulphite reaction. The blue colour denotes ‘T’ (thymine) corresponding to unmethylated cytosine, which is converted to uracil in the bisulfite reaction, and subsequently amplified to thymine during PCR. c, Methylation level at CpG sites in ALKATI-expressing tumour samples (MM-15 and ATC-28) and non-ALKATI-expressing control cells (H3122, a lung cancer cell line with EML4–ALK expression and SKMEL-28, a melanoma cell line without ALKATI expression). d, Comparison of the methylation status of CpG sites adjacent to the ATI site in ALKATI-expressing tumour samples (MM-15 and ATC-28) and non-ALKATI-expressing control cells (H3122 and SKMEL-28). The regions flanking LTR16B2 have significantly lower CpG methylation levels in ALKATI-expressing samples than controls; red dots indicate a statistically significant difference (P < 0.05; Mann–Whitney test) between ALKATI-expressing and non-expressing samples. Black dots indicate no statistically significant difference. e, ChIP-seq profile of H3K27ac at the ALKATI locus. The 17 blue profiles were retrieved from ENCODE, the 5 red profiles are original data from our lab. Only the 3 melanoma samples (MM-15, SKMEL-28, and SKMEL-524; bottom), but not the 19 non-melanoma cell lines, show H3K27ac enrichment at the ATI site. f, ChIP–qPCR validation for the H3K27ac enrichment at the ATI site in 6 melanoma cell lines. Error bars show mean ± s.e.m.; n = 3 technical replicates. g, Luciferase reporter assay of LTR16B2 in melanoma cell lines (red) and lung cancer cell lines expressing EML4–ALK (green). Error bars show mean ± s.d.; n = 9 (3 biological replicates combined from 3 independent experiments).
Extended Data Figure 7 ALKATI is active in vitro, shows nuclear and cytoplasmic localization by immunohistochemistry, and induces tumorigenesis.
a, In vitro kinase assay. The indicated ALK variants were stably expressed in NIH-3T3 cells, immunoprecipitated, and assayed for tyrosine kinase activity. After the enzymatic reaction, the immunoprecipitated material was used for immunoblots to assess the amount of ALK protein used in the kinase assay. Error bars, mean ± s.d.; n = 4 technical replicates. b, Melanoma (MM-15) expressing ALKATI shows cytoplasmic and nuclear localization of ALK by immunohistochemistry. Melanocytic tumour expressing a TPM3–ALK translocation shows a cytoplasmic localization of the ALK fusion protein. Fibroblasts, epithelial cells, and reactive lymphocytes serve as internal negative controls. Scale bars, 100 µm. c, Flow cytometry analysis for green fluorescent protein (GFP) co-expressed from the same ALK-expression vector. Cells were cultured in IL-3-supplemented medium until day 0 (blue curve) and the number of GFP-positive cells was assessed. The number of GFP-positive ALK-expressing cells was assessed again 14 days after IL-3 withdrawal (red curve). d, Immunoblots of explanted NIH-3T3 tumour grafts expressing the indicated ALK isoforms. ALKATI was expressed at similar protein levels as in two ALKATI-expressing clinical human tumour samples. e, Growth curves of tumour grafts of melan-a cells stably expressing the indicated ALK isoforms in cohorts of 4–5 mice each with bilateral grafts. Error bars, mean ± s.e.m.; n = 8 tumours for ALKF1174L, n = 10 tumours for all other experimental groups; see also Source Data associated with this figure. f, Immunoblots of explanted melan-a tumour grafts expressing the indicated ALK variants compared to ALKATI-expressing human tumour samples. g, Flow cytometry analysis of the GFP signal in NIH-3T3 cells stably expressing low (ALKATI-low) or high levels of ALKATI (ALKATI-high) before grafting into SCID mice. h, Immunoblot of t-ALK in ALKATI-low and ALKATI-high cells, confirming differential expression of ALKATI. See Supplementary Fig. 1 for uncropped blots for a, d, f and h. i, Growth curves of tumour grafts of ALKATI-low and ALKATI-high cells. Error bars, mean ± s.e.m.; n = 10 tumours; see also Source Data associated with this figure.
Extended Data Figure 8 Concentration-dependent ALK inhibition in ALKATI-, wild-type ALK-, ALKF1174L-, and EML4–ALK-expressing Ba/F3 cells.
a, b, Cell viability assay of Ba/F3 cells, either in the presence or absence of IL-3 (1 ng ml−1), expressing the indicated ALK isoforms and treated with the indicated doses of ALK inhibitors ceritinib (a) and TAE-684 (b). Cell viability was measured after 72 h of drug treatment. Error bars, mean ± s.e.m.; n = 3 biological replicates. c–e, Representative immunoblots of Ba/F3 cells stably expressing wild-type ALK (c), ALKF1174L (d), or EML4–ALK (e) and treated with increasing concentration of crizotinib for 2 h. See Supplementary Fig. 1 for uncropped blots.
Extended Data Figure 9 Expression of wild-type ALK, ALKF1174L, and EML4–ALK confers sensitivity to the ALK inhibitor crizotinib in vivo.
a–c, Bioluminescence of luciferase-labelled NIH-3T3 grafted tumours expressing wild-type ALK (a), ALKF1174L (b), or EML4–ALK (c) in SCID mice treated with either vehicle or crizotinib. Error bars, mean ± s.e.m.; n = 8 tumours; see also Source Data associated with this figure. d–f, H&E staining and immunohistochemistry of explanted tumours expressing wild-type ALK (d), ALKF1174L (e), or EML4–ALK (f) 48 h after the first crizotinib treatment. Scale bar, 50 μm. g, MSK-IMPACT assay reveals copy number alterations and loss of CDKN2A and PTEN in melanoma metastasis MM-382, but no mutations. The log2 ratio was calculated across all targeted regions by comparing the coverage in tumour versus normal. h, FISH for ALK shows no rearrangement; the 3 juxtaposed green/orange signals indicate a trisomy 2. Scale bar, 1 μm. i, The four FISH signals for MET and centromere 7 indicate a tetrasomy 7, but no MET amplification. Scale bar, 1 μm.
Supplementary information
Supplementary Figure 1
This file contains the uncropped blots for Figures 2b, 2c, 2d, 3b, 4b and Extended Data Figures 2a, 7a, 7d, 7h, 8c, 8d, 8e. (PDF 2346 kb)
Supplementary Table 1
This file contains percentage of reads starting at the ATI site in ALKATI-expressing tumour samples. (XLSX 39 kb)
Supplementary Table 2
This file contains single nucleotide polymorphisms and small indels at the ALK locus detected by ultra-deep sequencing. This file name was corrected on 2 November 2015. (XLSX 49 kb)
Supplementary Table 3
This file contains (a) single nucleotide polymorphisms and small indels in MM-15 detected by whole-genome sequencing, and (b) single nucleotide polymorphisms and small indels in ATC-28 detected by whole-genome sequencing. This file name was corrected on 2 November 2015. (XLSX 517 kb)
Supplementary Table 4
This file contains (4a) structural aberrations in MM-15 detected by whole-genome sequencing and (4b) structural aberrations in ATC-28 detected by whole-genome sequencing. (XLSX 1836 kb)
Supplementary Table 5
This file contains transcription factor motif analysis of the proximal cis-regulatory region (hg19 chr2: 29,445,000 to 29,447,100). (XLSX 52 kb)
Supplementary Table 6
This file contains the primer sequences used for ChIP-qPCR. (XLSX 37 kb)
Supplementary Table 7
The file contains probe set of Nanostring nCounter Assay to quantify mRNA levels of ALK variant transcripts in both fresh-frozen and formalin-fixed, paraffin-embedded (FFPE) specimens. (XLSX 53 kb)
Rights and permissions
About this article

Cite this article
Wiesner, T., Lee, W., Obenauf, A. et al. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature 526, 453–457 (2015). https://doi.org/10.1038/nature15258
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature15258
This article is cited by
-
Activation of human endogenous retroviruses and its physiological consequences
Nature Reviews Molecular Cell Biology (2024)
-
Multi-omic and functional analysis for classification and treatment of sarcomas with FUS-TFCP2 or EWSR1-TFCP2 fusions
Nature Communications (2024)
-
Pan-cancer analysis identifies tumor-specific antigens derived from transposable elements
Nature Genetics (2023)
-
Dominant role of DNA methylation over H3K9me3 for IAP silencing in endoderm
Nature Communications (2022)
-
CRISPRi enables isoform-specific loss-of-function screens and identification of gastric cancer-specific isoform dependencies
Genome Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
