
Abstract
Angiotensin converting enzyme (ACE) inhibitors reduce left ventricular (LV) hypertrophy and cardiovascular–renal fibrosis. Experimentally, changes in the LV and kidney persist even after cessation of treatment. The present study investigates whether brief ACE inhibition in spontaneously hypertensive rats (SHR) provides long-term protection against the LV and kidney damage induced by the nitric oxide synthase inhibitor N-ω-nitro-L-arginine-methyl ester (L-NAME). SHR received the ACE inhibitor enalapril (n=36) or tap water (n=36). In all, 12 control and treated SHR were sacrificed after 2 weeks and remaining rats were taken off-treatment. After a 2-week washout, 12 controls or previously treated SHR were sacrificed and remaining rats were treated with L-NAME ((control (Con)+L, enalapril (Enal)+L) for 10 days. At sacrifice, blood pressure was recorded via carotid artery cannulation in anesthetized rats, and blood, the kidney and LV were isolated for analysis. LV mass and arterial pressure were significantly reduced by enalapril. LV mass showed a persistent reduction throughout the study. In LV, prior enalapril treatment provided significant (P<0.05) protection against L-NAME-induced increases in proliferating cells (Con+L: 11±10.0 mm2 vs. Enal+L: 4±4.4 mm2), interstitial fibrosis (Con+L: 3±2.5% vs. Enal+L: 1±1.0%) and tissue macrophages (Con+L: 12±9 mm2 vs. Enal+L: 5±3.6 mm2). In the kidney, prior enalapril treatment protected against L-NAME-induced interstitial fibrosis and vascular injury. There was no difference in glomerular size or glomerulosclerosis regardless of prior treatment. Plasma creatinine and urea were significantly increased in L-NAME treated rats. This study suggests that brief ACE inhibition confers protection against future heart and kidney injury, even in the absence of continued antihypertensive treatment.
Similar content being viewed by others
Introduction
Despite increasing pharmacological choices, cardiovascular disease remains the leading cause of morbidity and mortality in North America with hypertension as the major risk factor in the majority of cardiovascular-related deaths.1 Continued research into the underlying mechanisms of disease and treatment paradigms should ultimately reduce or prevent the damaging effects to target organs including the heart and kidney. Certain antihypertensive drugs have been shown to provide long-term benefits beyond blood pressure lowering. For example, angiotensin converting enzyme (ACE) inhibitors have been shown clinically2 and experimentally,3, 4 to reduce cardiovascular mortality, in part by decreasing cardiac and vascular hypertrophy5 as well as cardiac and renal fibrosis.6, 7, 8 We have previously demonstrated in experimental models of hypertension that ACE inhibitor-induced regression of cardiac hypertrophy is mediated in part by apoptosis of fibroblasts in addition to reduction in cardiomyocyte size in the left ventricle (LV).5 Interestingly, this reduction in LV mass as well as arterial pressure have been shown to persist even after treatment cessation.9 Furthermore, short-term ACE inhibition has also produced kidney-specific changes including reduced renal structurally-based vascular resistance properties that persist long after stopping treatment.10
In animal models of myocardial infarction, initiating treatment with an ACE inhibitor before and during an ischemic insult results in a reduction in both infarct size and suppression of the inflammatory response.11, 12 Furthermore, concomitant ACE inhibitor treatment has also been shown to be protective against nitric oxide synthase (NOS) inhibitor (for example, N-ω-nitro-L-arginine-methyl ester (L-NAME))-induced LV fibrosis.13, 14 Although continued treatment with ACE inhibitors provides protection against target organ damage, the mechanisms responsible for these beneficial effects have not been fully elucidated. Thus, the present study sought to determine if transient ACE inhibition may confer some degree of protection against the subsequent development of cardiac and renal fibrosis induced by L-NAME administration.
Methods
Animal procedures
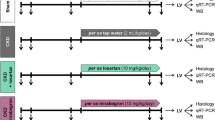
Young adult (11-weeks old) male spontaneously hypertensive rats (SHR) (Charles River, St-Constant, Quebec, Canada) were housed for at least 5 days before the initiation of treatment with either placebo (tap water, n=36) or the ACE inhibitor enalapril (30 mg kg−1 per day, P.O. n=36), as outlined in Figure 1. At the end of the 2-week treatment, a subset of 12 rats from each group was sacrificed (control (Con), enalapril (Enal)). All rats were then provided with tap water for a 2-week washout period. At the end of this period, a subset of rats was sacrificed (Con-off, Enal-off; n=12 per group, Figure 1). The remaining rats were then all treated with the NOS inhibitor, L-NAME (15 mg kg−1 per day, P.O.) for 10 days (Con+L, Enal+L; n=12 per group, Figure 1). Rats of similar body weight were pair-housed and all drugs were administered in the drinking water. Concentrations were determined based on average body weight and water consumption per cage, and adjusted every 5–6 days to account for changes in body weight and daily water consumption, as previously described.9, 10 Rats had ad libitum access to food and water throughout the treatment period. The dose and duration of enalapril treatment was based on previous studies where we demonstrated persistent reductions in blood pressure and LV mass following cessation of treatment9 and apoptosis-mediated reduction of cardiac fibrobasts in the LV.5 A 14-day washout period following enalapril treatment based on previous assessments demonstrating that blood pressure stabilizes at a new baseline ∼7–10 days following cessation of treatment.9, 10 The dose and duration of L-NAME was based on our previous unpublished observations demonstrating a marked induction of cardiac fibrosis.

Schematic representation of treatment design. Rats were initially divided into two treatment groups: control (Con) and enalapril (Enal). Following a 2-week treatment, the first set of rats (Con 2 week, Enal 2 week; n=12 per group) was sacrificed whereas the remaining animals were taken off-treatment. At the end of the 2-week washout period, the second sets were sacrificed (Con-off, Enal-off; n=12 per group) and L-NAME treatment was initiated in the remaining animals. After 10 days, the final sets of SHR were sacrificed (Con+L, Enal+L; n=12 per group).
At the time of sacrifice, rats were anesthetized (sodium pentobarbital 60 mg kg−1) and the left carotid artery cannulated for blood pressure measurement using a pressure transducer (Harvard Apparatus, St Laurent, QC, Canada) and a digital data acquisition system (Model MP100, Biopac System, Harvard Apparatus). Following blood pressure assessment, blood was harvested in sodium citrate containing tubes for plasma creatinine and urea assessments. An intravenous injection of cadmium chloride was then administered to induce diastolic cardiac arrest. Hearts were excised and left and right ventricles were separated, blotted dry and weighed. A transverse slice was taken from the middle of the LV and formalin fixed (TissuFix, Chaptec Inc, Montreal, QC, Canada) for histology. The right kidney was weighed and fixed for histological analysis. All procedures were in accordance with the Canadian Council on Animal Care.
Immunohistochemistry
In separate sections (5 μm), primary antibody for ED-1 (to identify macrophages; Abd-Serotec, Raleigh, NC, USA), α-smooth muscle actin (to identify vascular smooth muscle cells and myofibroblasts; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or proliferating cell nuclear antigen (to identify proliferating cells)-HRP (Dako North America, Carpinteria, CA, USA) was applied for 1 h. Anti-mouse biotinylated secondary antibody (Dako), followed by streptavidin-HRP were used with antibodies to ED-1 and α-smooth muscle actin. Positive staining was detected by diaminobenzidine reaction with horseradish peroxidase resulting in a brown precipitate. Tissues were counterstained with hematoxylin to identify nuclei. Negative controls were performed without adding the primary antibody and did not display any brown precipitate. Total number of cells positively labeled for either ED-1 or proliferating cell nuclear antigen was determined in the entire LV section and normalized to cross-sectional area.
Cardiac and renal histology
The LV and kidney sections were stained with Gomori's trichrome stain (Newcomer Supply, Middleton, WI, USA) to identify collagen fibers. The area of perivascular fibrosis was normalized to coronary artery cross-sectional area—each of which was analyzed using Image J software (NIH). Evaluation of fibrosis in stained LV was determined at a × 400 magnification. A blinded observer determined the percentage area stained for collagen. Specifically, a 121-point grid was overlaid on an image and the points touching the blue stained collagen were counted. Every other field was counted until the entire LV was scanned (that is, 50% of the section was analyzed). The number of points of intersection with collagen stain was divided by the total number of potential intersection sites (for example, 121 × number of fields evaluated). Fields that contained large arteries with perivascular fibrosis were not evaluated. Separate sections were stained with picrosirius red for determination of coronary artery medial wall fibrosis using Image J to evaluate percentage fibrosis per cross-sectional area.
Kidney sections were stained with periodic acid schiff to evaluate vascular injury. A full 1/2 kidney longitudinal section through hilum was made and the entire cortex was scored. The score itself was made by taking a count of number of vessels (or vessel and primary branch) affected by hyaline arteriolar changes or muscular wall thickening. Renal interstitial fibrosis was evaluated using the same approach in sections stained with Gomori's Trichrome stain and counting cortical scars. A single scar was defined as any area of interstitial fibrosis >0.1 mm up to one × 20 field (0.93 mm), with larger areas counted as two scars. Glomerular assessments were performed in sections stained with sirius red as described previously.15 Each section was examined by a kidney pathologist (SJR) in a blinded manner with regards to treatment groups.
Statistics
When the data from only two groups were being compared, an unpaired Student's t-test was employed. Comparisons involving more than two groups were made using a one-way ANOVA with a Bonferroni post-hoc test for multiple comparisons (Graph Pad, La Jolla, CA, USA). Data are expressed as mean±s.d. and differences were deemed statistically significant with a P<0.05.
Results
Blood pressure and cardiac hypertrophy
Body weight was not different across treatment groups (Table 1). Arterial pressure was significantly reduced by enalapril after 2 weeks; however, this difference did not remain statistically significant in the off-treatment period (Table 1). L-NAME treatment significantly increased arterial pressure to a similar extent regardless of prior treatment. In addition, plasma urea and creatinine were increased in all SHR (Table 1).
LV mass relative to body weight was significantly reduced by enalapril and this difference persisted throughout the treatment periods. Following 10 days of L-NAME treatment, LV mass relative to body weight was significantly increased by an equivalent magnitude in both groups (Table 1). Right ventricle mass relative to body weight was not modified by enalapril treatment; however, L-NAME-induced RV hypertrophy was lower in SHR previously treated with enalapril (Table 1). Kidney weight relative to body weight was not modified by any treatment (Table 1).
Cardiac histology
Fibrosis was quantified in the ‘inner’ (sub-endocardial) and ‘outer’ (sub-epicardial plus mid-myocardial) regions of the SHR LV in the off-treatment period, as well as following L-NAME challenge (Figures 2a and b). The distinction was based on histological examination of cardiomyocytes. For example, the ‘inner’ (sub-endocardium) region was defined based on cross-sectional appearance of cardiomyocytes (as in papillary muscle). This region of the LV myocardium displayed a relatively low level of fibrosis that was not modified significantly by any treatments (Figure 2c). The degree of fibrosis in the outer LV myocardium was not different between previously treated enalapril rats (Enal-off) and age-matched control (Con-off) (Figure 2d). However, there was a marked increase in collagen accumulation with L-NAME in SHR that were not previously treated (Con+L) (Figure 2d). The nature of the cardiac pathology generally suggested a loss of cardiomyocytes followed by ‘replacement fibrosis’. In contrast, the degree of cardiac fibrosis in rats previously treated with enalapril (Enal+L) was not modified by L-NAME (Figure 2d). Perivascular fibrosis of coronary arteries was measured in L-NAME treated rats (Figures 2e and f). There was no difference in the area of perivascular fibrosis when normalized to artery cross-sectional area (Figure 2g), nor was there an impact on medial wall fibrosis (Figure 2h).

Prior enalapril treatment significantly prevented L-NAME induced fibrosis. Representative images from Gomori trichrome stained Con+L (a) and Enal+L (b) LVs taken at × 100 magnification. There was no change in collagen deposition in the inner wall (c). In the outer wall, L-NAME significantly increased collagen deposition in previously untreated rats. In contrast, Enal+L hearts had significantly less extracellular matrix deposition than Con+L, and was not different from Enal-off (d). Representative images of coronary arteries from Con+L (e) and Enal+L (f) taken at × 400 magnification. Area of perivascular fibrosis was not modified when normalized for vessel cross-sectional area (g), whereas there was a trend towards reduced medial wall collagen deposition (h). †P<0.05 vs. Con-off, *P<0.05 vs. Con+L. Bar represents 500 μm. A full color version of this figure is available at the Hypertension Research journal online.
Immunohistochemical analysis was performed in LV of L-NAME treated rats to evaluate changes in macrophage infiltration (ED-1-positive cells, Figures 3a top and b), proliferating cells (proliferating cell nuclear antigen-positive cells, Figures 3a middle and c) and alpha smooth muscle actin-positive cells (Figure 3a bottom). Proliferating cell nuclear antigen-positive and ED-1-positive cells were present throughout the myocardium, but tended to cluster in the perivascular and pericardial regions as well as areas of active remodeling—regions that had an increased proportion of non-cardiomyocytes and extracellular matrix (Figure 3a). Alpha smooth muscle actin-positive cells, suggesting a potential transition of fibroblast to myofibroblast phenotype, were located in areas of active remodeling, but were generally less apparent in the perivascular region (Figure 3a). These cells were distinguished from vascular smooth muscle cells if they were located in an area distinct from a vessel (Figure 3a). ED-1-positive macrophages were observed in all LV from L-NAME treated rats, with a significantly greater number in the Con+L hearts, compared with the SHR previously treated with enalapril (Figure 3b). Proliferating cells were observed in LV from all L-NAME treated rats, with a significantly greater number of cells in the Con+L group, compared with SHR previously treated with enalapril (Figure 3c). Given the nature and extent of the α-smooth muscle actin labeling it was difficult to quantify this cell type and therefore this parameter was only evaluated qualitatively. (Colour images are available in HTML version.)

Prior enalapril treatment markedly reduced the degree of macrophage infiltration and number of proliferating cells in response to L-NAME treatment. All representative images (a) were taken from Con+L hearts at × 400 magnification to demonstrate the location of ED-1-positive (brown stain, arrows) macrophages (a, top), proliferating cell nuclear antigen (PCNA)-positive (brown stain, arrows) cells (a, middle) and α-smooth muscle actin-positive (brown stain) cells (a, bottom). Quantification of macrophages per unit area (b) revealed a significant reduction in the LV from Enal+L compared with Con+L SHR. Quantification of PCNA-positive cells per unit area (c) revealed a significant reduction in the LV from Enal+L compared with Con+L SHR. *P<0.05 vs. Con+L. Bar represents 200 μm. A full color version of this figure is available at the Hypertension Research journal online.
Renal histology
In L-NAME-treated SHR, there was minimal evidence of glomerulosclerosis (Con+L 0.1±0. 063% vs. Enal+L 0.1±0.034%) or glomerular hypertrophy (Con+L 0.13±0.006 mm vs. Enal+L 0.13±0.004 mm), regardless of prior treatment. Interstitial fibrosis was not evident in the off-treatment rats; however, it was markedly increased following L-NAME treatment (Figures 4a and e). In contrast, the L-NAME-induced renal interstitial fibrosis was markedly reduced in SHR that were previously treated with enalapril (Figures 4b and e). In the off-treatment period there was minimal evidence of vascular injury. However, L-NAME treatment produced a significant increase in the histological score compared with rats that did not receive L-NAME (Figures 4c and f). As with the cardiac and renal fibrosis, the vascular damage was significantly reduced in the Enal+L group (Figures 4d and f).

Prior enalapril treatment mitigated L-NAME induced vascular injury and interstitial fibrosis. Representative images of kidneys stained with Gomori Trichrome from Con+L (a) and Enal+L (b) SHR taken at × 40 magnification. Con+L (a) kidney displays marked interstitial scarring (blue stain) and tubular atrophy (arrows). Representative images ( × 200 magnification) from periodic acid schiff stained kidneys from Con+L (c) and Enal+L (d) SHR. L-NAME treatment caused a marked increase in renal vascular injury in Con+L (c), as evidenced by hyaline changes in arterioles (black arrow) and muscular hypertrophy (white arrows), with minimal changes observed in Enal+L kidney (d). Quantified scores for interstitial fibrosis (e) and vascular injury (f) revealed significant differences between groups. †P<0.05 vs. Con-off, *P<0.05 vs. Con+L, ‡P<0.05 vs. Enal-off. A full color version of this figure is available at the Hypertension Research journal online.
Discussion
The major findings of the present study were that in the face of NOS inhibition, those rats that previously received enalapril treatment displayed a resistance to development of cardiac fibrosis, inflammatory cell infiltration and cellular proliferation as well as renal pathological remodeling involving fibrosis and vascular injury. Importantly, the hemodynamic challenge was equivalent in previously treated and untreated groups. Furthermore, this protection was observed without ongoing antihypertensive treatment. That is, short-term ACE inhibition produced a phenotypic change in the LV and kidney of adult SHR that persisted into the off-treatment period and reduced the susceptibility to the pathological remodeling that underlies heart failure and kidney disease.
It has long been known that treatment of young SHR in the pre-hypertensive phase with ACE inhibitors, but not direct acting vasodilators, prevents the development of hypertension and cardiovascular hypertrophy and that this ‘normotensive’ phenotype persists long after cessation of treatment.16, 17, 18, 19 Later, it was determined that short-term (2-week) ACE inhibition in adult SHR was capable of reversing established hypertension and cardiovascular hypertrophy and that this ‘normotensive’ phenotype also persists long after cessation of treatment.9, 10 Recently Ishiguro et al.16 used the former paradigm and demonstrated that by preventing development of hypertension, the adult SHR had a reduced susceptibility to L-NAME-induced pathological remodeling of the kidney. The suggestion was that if hypertension is never able to develop, the rats would be resistant to the damaging effects of NOS inhibition. In separate studies, a 7-week angiotensin receptor blocker treatment in pre-pubertal rats protected against future development of diabetic nephropathy.20 In each of these studies, the authors concluded that treatment during a ‘critical period’ during development would confer protection against future injury.16, 20 Importantly the present study demonstrates for the first time that initiating a 2-week treatment in adult SHR with fully established hypertension and cardiovascular hypertrophy could confer protection against a future insult in both the heart and kidney. Thus, the critical period for developing drug-induced protection against future injury may not be restricted to the young age.
We now report that after only 10 days, L-NAME-induced fibrosis develops predominantly in the outer myocardium. Although an age-matched control SHR was not included, it is likely that the marked increase in LV fibrosis is because of L-NAME and not a natural progression over the 10-day span given that SHR of similar age do not display a high level of cardiac collagen deposition. For example, Ahn et al.21 demonstrated that between the ages of 8 and 20 weeks there was not a marked increase in hydroxyproline content in the SHR LV. Remarkably, there was no increase in LV collagen content in animals previously treated with enalapril. Given the nature of the fibrosis, this may suggest that prior enalapril treatment protected against cardiomyocyte loss, however, this will need to be tested directly. We have previously demonstrated that a 2-week treatment with enalapril induced a transient wave of apoptosis resulting in a 30% reduction in cardiac fibroblasts, an event that occurred mainly in the outer myocardium.5 It may be that the prior enalapril treatment removed a subset of fibroblasts that are largely responsible for the excessive collagen deposition in the Con+L rats. Although this hypothesis remains to be fully explored, a reduction or modulation of the fibroblast population could potentially limit the fibrotic response, given this cell's role in maintaining the extracellular matrix composition. The pro-fibrotic effects are thought to be mediated by a differentiated and activated fibroblast phenotype termed ‘myofibroblast’. These cells are typically characterized by expression of α-smooth muscle actin, embryonic smooth muscle myosin, fibronectin ED-A and fibrillar collagens; and exhibit increased migratory, proliferative and secretory properties.22 Myofibroblasts are not only responsive to but also actively secrete growth factors, proteases, ECM components and pro-inflammatory cytokines during active phases of cardiac tissue repair and fibrosis.22
Evidence is accumulating that suggests that inflammation may have a critical role in the development of fibrosis. Furthermore, fibroblasts, once believed to serve only as producers of collagen, may have a critical role in mediating the inflammatory response to injury.23 Fibroblasts may mediate the switch from acute inflammatory response to a situation of chronic inflammation.24 Mast cells,25 macrophages26 and T cells27 have all been found at the site of active remodeling and cytokines released from these cells can promote collagen deposition and fibroblast proliferation. In animal models of pressure overload-induced LV hypertrophy, a transient increase in cytokine release and macrophage infiltration28 has been shown, and Hsieh et al.29 have demonstrated that NOS inhibition with L-NAME in hypertensive rats increases vascular inflammation. Although previous studies have demonstrated that ACE inhibitor treatment reduces macrophage infiltration and cellular proliferation following myocardial infarction,30 the present study is the first to demonstrate that these beneficial effects persist well after stopping treatment. Specifically, there was a >50% reduction in the number of macrophages in the LV of SHR previously treated with enalapril. It may be that the prior removal of 30% of cardiac fibroblasts that has been previously shown to occur during ACE inhibitor treatment,5, 31 results in a modified immune response in the early stages of L-NAME-induced pathological remodeling. By reducing or changing the fibroblast population, the immune response may therefore have been more appropriate in limiting the excessive scarring in response to NOS inhibition.
The findings from the present study demonstrate that short-term ACE inhibitor treatment has long-term effects in preventing cardiac and renal injury induced by nitric oxide deficiency. Interestingly, this suggests that an animal genetically predisposed to target organ damage can be modified such that the response to a pathological challenge is reduced. Future work emanating from these studies may reveal greater understanding of the mechanisms of cardiac and renal protection afforded by ACE inhibitor treatment, as well as reveal novel treatment strategies for the treatment or prevention of fibrotic diseases.
References
Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De SG, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger Mussolino M, Nichol VL, Rosamond W, Sacco R, Sorlie P, Roger VL, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J . Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 2010; 121: e46–e215.
Arnold JM, Yusuf S, Young J, Mathew J, Johnstone D, Avezum A, Lonn E, Pogue J, Bosch J . Prevention of Heart Failure in Patients in the Heart Outcomes Prevention Evaluation (HOPE) Study. Circulation 2003; 107: 1284–1290.
deBlois D, Tea BS, Than VD, Tremblay J, Hamet P . Smooth muscle apoptosis during vascular regression in spontaneously hypertensive rats. Hypertension 1997; 29: 340–349.
Schiffrin EL, Deng LY, Larochelle P . Effects of antihypertensive treatment on vascular remodeling in essential hypertensive patients. J Cardiovasc Pharmacol 1994; 24 (Suppl 3): S51–S56.
Der Sarkissian S, Marchand EL, Duguay D, Hamet P, deBlois D . Reversal of interstitial fibroblast hyperplasia via apoptosis in hypertensive rat heart with valsartan or enalapril. Cardiovasc Res 2003; 57: 775–783.
Perret-Guillaume C, Joly L, Jankowski P, Benetos A . Benefits of the RAS blockade: clinical evidence before the ONTARGET study. J Hypertens Suppl 2009; 27: S3–S7.
Susic D, Varagic J, Frohlich ED . Pharmacologic agents on cardiovascular mass, coronary dynamics and collagen in aged spontaneously hypertensive rats. J Hypertens 1999; 17: 1209–1215.
Liang B, Leenen FH . Prevention of salt induced hypertension and fibrosis by angiotensin converting enzyme inhibitors in Dahl S rats. Br J Pharmacol 2007; 152: 903–914.
Hale TM, Shoichet MJ, Bushfield TL, Adams MA . Time course of vascular structural changes during and after short-term antihypertensive treatment. Hypertension 2003; 42: 171–176.
Smallegange C, Hale TM, Bushfield TL, Adams MA . Persistent lowering of pressure by transplanting kidneys from adult spontaneously hypertensive rats treated with brief antihypertensive therapy. Hypertension 2004; 44: 89–94.
Zhu B, Sun Y, Sievers RE, Browne AE, Pulukurthy S, Sudhir K, Lee RJ, Chou TM, Chatterjee K, Parmley WW . Comparative effects of pretreatment with captopril and losartan on cardiovascular protection in a rat model of ischemia-reperfusion. J Am Coll Cardiol 2000; 35: 787–795.
Weidenbach R, Schulz R, Gres P, Behrends M, Post H, Heusch G . Enhanced reduction of myocardial infarct size by combined ACE inhibition and AT(1)-receptor antagonism. Br J Pharmacol 2000; 131: 138–144.
Pereira LM, Bezerra DG, Machado DL, Mandarim-de-Lacerda CA . Enalapril attenuates cardiorenal damage in nitric-oxide-deficient spontaneously hypertensive rats. Clin Sci (Lond) 2004; 106: 337–343.
Katoh M, Egashira K, Mitsui T, Chishima S, Takeshita A, Narita H . Angiotensin-converting enzyme inhibitor prevents plasminogen activator inhibitor-1 expression in a rat model with cardiovascular remodeling induced by chronic inhibition of nitric oxide synthesis. J Mol Cell Cardiol 2000; 32: 73–83.
Dilauro M, Zimpelmann J, Robertson SJ, Genest D, Burns KD . Effect of ACE2 and angiotensin-(1–7) in a mouse model of early chronic kidney disease. Am J Physiol Renal Physiol 2010; 298: F1523–F1532.
Ishiguro K, Sasamura H, Sakamaki Y, Itoh H, Saruta T . Developmental activity of the renin-angiotensin system during the ″critical period″ modulates later L-NAME-induced hypertension and renal injury. Hypertens Res 2007; 30: 63–75.
Adams MA, Bobik A, Korner PI . Enalapril can prevent vascular amplifier development in spontaneously hypertensive rats. Hypertension 1990; 16: 252–260.
Wu JN, Berecek KH . Prevention of genetic hypertension by early treatment of spontaneously hypertensive rats with the angiotensin converting enzyme inhibitor captopril. Hypertension 1993; 22: 139–146.
Christensen KL, Jespersen LT, Mulvany MJ . Development of blood pressure in spontaneously hypertensive rats after withdrawal of long-term treatment related to vascular structure. J Hypertens 1989; 7: 83–90.
Ishiguro K, Sasamura H, Sakamaki Y, Hayashi K, Saruta T, Itoh H . Differential effects of transient treatment of spontaneously hypertensive rats with various antihypertensive agents on the subsequent development of diabetic nephropathy. Nephron Exp Nephrol 2008; 109: e20–e28.
Ahn J, Varagic J, Slama M, Susic D, Frohlich ED . Cardiac structural and functional responses to salt loading in SHR. Am J Physiol Heart Circ Physiol 2004; 287: H767–H772.
Porter KE, Turner NA . Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther 2009; 123: 255–278.
Smith RS, Smith TJ, Blieden TM, Phipps RP . Fibroblasts as sentinel cells. Synthesis of chemokines and regulation of inflammation. Am J Pathol 1997; 151: 317–322.
Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M . Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol 2001; 22: 199–204.
Levick SP, McLarty JL, Murray DB, Freeman RM, Carver WE, Brower GL . Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension 2009; 53: 1041–1047.
Hamid T, Gu Y, Ortines RV, Bhattacharya C, Wang G, Xuan YT, Prabhu SD . Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation. Circulation 2009; 119: 1386–1397.
Yu Q, Horak K, Larson DF . Role of T lymphocytes in hypertension-induced cardiac extracellular matrix remodeling. Hypertension 2006; 48: 98–104.
Xia Y, Lee K, Li N, Corbett D, Mendoza L, Frangogiannis NG . Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem Cell Biol 2009; 131: 471–481.
Hsieh NK, Wang JY, Liu JC, Wang SD, Chen HI . Nitric oxide inhibition accelerates hypertension and induces perivascular inflammation in rats. Clin Exp Pharmacol Physiol 2004; 31: 212–218.
Peng H, Carretero OA, Vuljaj N, Liao TD, Motivala A, Peterson EL, Rhaleb NE . Angiotensin-converting enzyme inhibitors: a new mechanism of action. Circulation 2005; 112: 2436–2445.
Duguay D, Sarkissian SD, Kouz R, Ongali B, Couture R, deBlois D . Kinin B2 receptor is not involved in enalapril-induced apoptosis and regression of hypertrophy in spontaneously hypertensive rat aorta: possible role of B1 receptor. Br J Pharmacol 2004; 141: 728–736.
Acknowledgements
This work was funded by a grant from the Canadian Institutes of Health Research to DD. TMH received a postdoctoral fellowship from the Heart & Stroke Foundation of Canada.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hale, T., Robertson, S., Burns, K. et al. Short-term ACE inhibition confers long-term protection against target organ damage. Hypertens Res 35, 604–610 (2012). https://doi.org/10.1038/hr.2012.2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2012.2
