
Abstract
Chromosomal inversions are common in natural populations and are believed to be involved in many important evolutionary phenomena, including speciation, the evolution of sex chromosomes and local adaptation. While recent advances in sequencing and genotyping methods are leading to rapidly increasing amounts of genome-wide sequence data that reveal interesting patterns of genetic variation within inverted regions, efficient simulation methods to study these patterns are largely missing. In this work, we extend the sequential Markovian coalescent, an approximation to the coalescent with recombination, to include the effects of polymorphic inversions on patterns of recombination. Results show that our algorithm is fast, memory-efficient and accurate, making it feasible to simulate large inversions in large populations for the first time. The SMC algorithm enables studies of patterns of genetic variation (for example, linkage disequilibria) and tests of hypotheses (using simulation-based approaches) that were previously intractable.
Similar content being viewed by others
Introduction
Inversions are chromosome mutations in which a chromosome breaks at two points and is reinserted in reversed orientation. Chromosomal inversions occur frequently as fixed differences between sister species (Ranz et al., 2007) and as polymorphisms within many populations (Dobzhansky, 1970). There is growing evidence that inversions are involved in many evolutionary phenomena such as local adaptation (Kennington et al., 2006), speciation (Bush et al., 1977; White, 1978), and the evolution of sex chromosomes (Charlesworth et al., 2005). Consequently, understanding how and why inversions evolve is an important problem in evolutionary biology (Hoffmann and Rieseberg, 2008; Kirkpatrick, 2010).
A key feature of inversions is that they alter recombination (Roberts, 1976). Recombination in chromosomal homozygotes (homokaryotypes) remains unaltered. Genetic exchange in chromosomal heterozygotes (heterokaryotypes), on the other hand, is greatly suppressed. Multiple crossovers (Ashburner, 1989) and gene conversion (Chovnick, 1973) can lead to viable recombinant gametes and hence to exchange of genetic material between chromosome arrangements. (These processes are, however, often neglected in theoretical studies, for example, Chen et al., 2006; O’Reilly et al., 2010.) Movement of genetic material by recombination between chromosomal arrangements is called ‘gene flux’ (Navarro et al., 1997). Flux is usually more strongly reduced for sites close to the breakpoints than it is near the center of the inversion (Novitski and Braver, 1954). Estimates for the rate of gene flux per nucleotide and per generation range from from 10−4 in the central region of inversion In(3L)Payne of Drosophila melanogaster to 10−7 near the breakpoints of heterokaryotypes in D. subobscura (Navarro et al., 2000).
Like other mutations, inversions can be neutral or selected. An intriguing example of selection is the cline in the well-studied inversion (3R)Payne in D. melanogaster (for example, Kennington et al., 2006). Direct selection on inversions can be caused by several mechanisms (reviewed in Kirkpatrick, 2010). Selection can also be indirect, induced by the inversion’s effect on recombination. This situation arises when the inversion carries one or more selected alleles (Dobzhansky, 1970; Kirkpatrick and Barton, 2006; Joron et al., 2011). Inversions are expected to be hotspots for locally adapted alleles, both because those alleles can cause an inversion to become established (Kirkpatrick and Barton, 2006) and because they differentially accumulate within inverted regions after an inversion is established (Navarro and Barton, 2003).
Because of their effects on recombination, inversions have significant effects on patterns of neutral genetic variation. These patterns carry information about an inversion’s history and the type of selection that it experiences (Kirkpatrick and Kern, 2012). Navarro et al., 2000 modeled inversions as balanced polymorphism and concluded that the effect of inversions on nucleotide variability depends mainly on the pattern of recombination and the age of the inversion. Neutral genetic divergence between chromosome classes is expected to be high at sites close to the breakpoints, whereas divergence should decay relatively quickly in the center of the inversion. In fact, observed levels of neutral genetic divergence often decrease from the inversion breakpoints towards its center (Navarro et al., 1997; Laayouni et al., 2003; Cheng et al., 2011; McGaugh and Noor, 2012). In Drosophila, linkage disequilibrium (LD) between the inversion and markers in the center of the inversion is expected to decrease to low levels in tens of thousands of generations (Andolfatto et al., 2001). A young inversion is expected to be in much stronger LD with markers inside the inversion. A recent study based on coalescent theory (Guerrero et al., 2012) showed that different models for the evolution of inversions leave in fact quite different genetic footprints, which opens the door for quantitative tests of competing hypotheses.
Recent advances in sequencing and genotyping methods provide an increasing number of data sets that reveal interesting patterns of neutral genetic variation within inverted regions (for example, Cheng et al., 2011; McGaugh and Noor, 2012). In principle, these patterns could point to genes under selection and be used to test alternative theories for how inversions evolve. Interpretation of the data is challenging; however, because they result from complex and interacting forces, including demography, population structure, recombination and selection.
The coalescent approach has proven to be a powerful tool to analyze patterns of genetic variation (for example, Hudson, 1983, 2002; Griffiths and Marjoram, 1996). Coalescent models that include recombination have been used successfully to analyze short chromosomal regions that lack structural variation such as inversions (Hudson, 2002; Laval and Excoffier, 2004; Ewing and Hermisson, 2010). Classical statistical inference based on likelihood is technically challenging, however, and for some evolutionary models, it is simply not possible (McVean and Cardin, 2005). Those constraints motivate likelihood-free inference methods such as approximate Bayesian computation, or ABC (Beaumont, 2010). This approach requires simulating very large numbers of genealogies under the hypotheses of interest. For chromosome segments, a natural way to represent the genealogy is with the ancestral recombination graph, or ARG (for example, Hudson, 1990; Griffiths and Marjoram, 1997). Standard methods for simulating the ARG of large chromosome segments in large populations, however, are not feasible because of computational limits.
One solution to this problem is to use an approximation to the coalescent model for which one can either calculate likelihoods or implement efficient simulations. In this work, we focus on the latter: finding an efficient and accurate algorithm for simulating the ancestral recombination graph (ARG) with inversions. It is based on the so-called sequential Markovian coalescent (SMC) that was proposed by McVean and Cardin, 2005 and later improved (Marjoram and Wall, 2006; Chen et al., 2009; Eriksson et al., 2009; Excoffier and Foll, 2011). Building on a recent coalescent approach for independent sites within an inversion (Guerrero et al., 2012), we here extend the SMC to allow for the non-standard patterns of recombination that result from inversions. We compare the algorithm to both forward-in-time and standard coalescent simulations and find that the algorithm leads to very accurate results. In addition, it is much faster and requires much less memory than exact simulations of the ARG. These findings suggest that the SMC algorithm with inversions could be an extremely useful tool to study neutral genetic variation in inversions.
Materials and Methods
The coalescent
Our work is based on the continuous-time coalescent (for example, Wakeley, 2008). The population consists of N diploid individuals that mate randomly. Time is measured in 2N generations. Let ρ=4Nr, where r is the map length of the stretch of DNA we want to simulate. In other words, r is the probability that a recombination event occurs in the region of interest in a single generation. It is convenient to scale the region of interest to the unit interval [0,1]. Let x ∈ [0, 1] denote the position on the chromosome. The coalescent tree at site x is as denoted T(x) and its total length is denoted L(x). We assume that all genealogical events occur infrequently, so we can ignore situations in which two events happen in the same generation. Let k be the number of ancestral lineages present at a point in time. The rate of coalescence between any two given lineages is 1, and the recombination rate for any lineage is ρ/2. Hence, coalescent or recombination events are exponentially distributed and occur at rates ( ) and kρ, respectively. In the case of a coalescent event, two randomly drawn lineages coalesce. In the case of a recombination event, a point in [0,1] is picked and a randomly drawn lineage splits in two branches. One branch corresponds to ancestral material to the left of the recombination point and the other to the material to the right. This process is continued until all loci have reached their most recent common ancestors. The resulting graph is called the ancestral recombination graph, or ARG (for example, Hudson, 1990; Griffiths and Marjoram, 1997).
) and kρ, respectively. In the case of a coalescent event, two randomly drawn lineages coalesce. In the case of a recombination event, a point in [0,1] is picked and a randomly drawn lineage splits in two branches. One branch corresponds to ancestral material to the left of the recombination point and the other to the material to the right. This process is continued until all loci have reached their most recent common ancestors. The resulting graph is called the ancestral recombination graph, or ARG (for example, Hudson, 1990; Griffiths and Marjoram, 1997).
SMC algorithm without inversions
The sequential Markovian coalescent (SMC) algorithm was introduced by McVean and Cardin (2005) as an approximation to the ARG based on an elegant scheme by Wiuf and Hein (1999). In this method, one moves along the chromosome and updates the ancestral recombination graph wherever a recombination event occurs. McVean and Cardin (2005) argued that ignoring coalescent events between lineages that do not share overlapping ancestral material has little impact on patterns of LD. Ignoring such events in the algorithm of Wiuf and Hein (1999) leads to the sequential Markovian coalescent. In essence, one moves along the chromosome and updates the coalescent tree, instead of the full ancestral recombination graph, whenever recombination occurs. This yields a coalescent tree for every position in the simulated region.
The standard SMC can be described as follows (cf. McVean and Cardin, 2005):
-
1
Construct a standard coalescent tree, that is, without recombination, at position x=0. The total length of the tree is L(x).
-
2
Pick the location of the next recombination event (moving along the chromosome). The distance to the next event, Δx, is exponentially distributed with parameter ρ/2L(x). If x+Δx>1, stop.
-
3
Pick a location on the tree (uniformly) and erase the part of the branch between the current position on the tree and the next coalescent event (moving backwards in time). In this step, a so-called floating lineage is created.
-
4
Go backwards in time from the current position on the tree. The floating lineage can coalesce with any of the remaining branches. The rate is proportional to the number of ancestral lineages present. After the floating lineage is reattached, go to step 2.
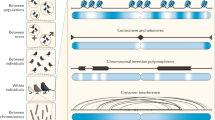
The algorithm is illustrated in Figure 1.

Sketch of the original SMC algorithm. In the first step, a recombination event is dropped on the tree (indicated by a crossmark). The next step shows how the upper part of the branch is removed and a floating lineage is created. In the last step, the new lineage (indicated by a bold line) is reattached to the tree.
It is straightforward to see that this algorithm preserves the original distribution of marginal genealogies. However, with increasing recombination between a pair of sites, the correlation between coalescence times decreases slightly faster than in the full ancestral recombination graph (McVean and Cardin, 2005). Nevertheless, the SMC has two big advantages. First, simulations using the SMC algorithm are much faster than simulations of the full ARG. Secondly, much less memory is required because at any given point in time, it is a single bifurcating tree that has to be stored instead of a potentially very large ancestral recombination graph. For a detailed discussion of this algorithm and its extensions we refer to the studies of McVean and Cardin (2005), Marjoram and Wall (2006), Chen et al. (2009), and Eriksson et al. (2009).
SMC with inversions
The presence of a polymorphic inversion necessitates changes in the SMC algorithm. The two main differences to the standard scenario are the following. First, the population is structured into two classes, standard and inverted, and only lineages belonging to the same class can coalesce. Second, inversions change the recombination pattern. In particular, recombination between heterokaryotypes is strongly reduced, whereas homokaryotypic recombination remains unaltered. The basic idea of our algorithm is to separate homokaryotypic recombination and gene flux. As in the traditional SMC algorithm, we simulate homokaryotypic recombination as we move along the chromosome. In contrast, gene flux is simulated as we construct the coalescent tree backwards in time. This is similar in spirit to an SMC algorithm that was recently developed for models with spatial structure (Chen et al., 2009; Eriksson et al., 2009).
We use a simple but general model for gene flux between the two chromosome classes. Exchange of genetic material between standard and inverted chromosomes occurs such that stretches of DNA move from one chromosome to the other. The flow of genes can be unidirectional, that is, genes from one chromosome are transferred to the other but not vice versa, or bidirectional, that is, the genetic material is exchanged between two chromosomes. These two cases correspond to gene flux resulting from gene conversion or double crossing-over, respectively. Let x ∈ [0, 1] denote the position on the chromosome, where 0 and 1 correspond to the breakpoints of the inversion and (0,1) is the inverted region. If gene flux occurs in a heterokaryotype, a subinterval I=(β1, β2) of (0,1) is moved from one chromosome to the other (see Figure 2 for a sketch of the model). The breakpoints β1 and β2 are random variables. We set ϕ(x)=Prob(β1<x<β2), that is, the probability that site x is affected by a (randomly drawn) gene flux event. We will call the events that occur at β1 and β2 the left and right part of a gene flux event respectively.

Sketch of how gene flux between different chromosome arrangements is modeled. The lines indicate a standard (black) and an inverted (gray) chromosome in a heterokaryotype. The letters indicate different sites along the chromosomes. The dashed lines indicate the boundaries β1 and β2 of the interval I that is moved from one chromosome to the other. The arrows symbolize that genetic material can be copied from one chromosome and inserted into the other (as in gene conversions), or that genetic material can be exchanged between the two chromosomes (as in double crossing-overs).
To construct a standard coalescent tree at a particular site x, we have to account for how gene flux changes the rate at which lineages coalesce. The total rate of gene flux per lineage can be written as  , where ph is the probability that the lineage finds itself in a heterokaryotype. We call c∈[0, 1] the gene flux coefficient; it measures the rate of gene flux in heterokaryotypes relative to the rate of recombination in homokaryotypes. The rate of gene flux at position x is then given by
, where ph is the probability that the lineage finds itself in a heterokaryotype. We call c∈[0, 1] the gene flux coefficient; it measures the rate of gene flux in heterokaryotypes relative to the rate of recombination in homokaryotypes. The rate of gene flux at position x is then given by

that is, the rate at which gene flux occurs in a single lineage times the probability that the current site is affected by it. For simplicity, we assume that the frequency of the inversion, pI, is constant at all times. (This assumption is not restrictive and the algorithm can be extended in a straightforward way to more complicated scenarios, for example, an inversion with a recent origin.) Going backwards in time, there are four possible events: coalescence of two inverted lineages, coalescence of two standard lineages, the ancestral material at x moves from an inverted chromosome to a standard chromosome or vice versa. The rates at which these events occur are

respectively, where kI or ks denotes the number of inverted or standard lineages respectively. The construction of the coalescent is then straightforward (cf. Guerrero et al., 2012).
It is important to observe that a gene flux event affects not only a single site but also neighboring sites. In particular, an interval (β1, β2) with β1<x<β2 is transferred from on chromosome to another. When and where this event occurs will be important in the sequential algorithm because the tree has to be updated at every recombination event as we move along the chromosome. In the ancestral recombination graph, a gene-flux event causes two recombination events; one at location β1 and one at β2. In the SMC, however, events to the left of the current position are ignored because we follow only lineages that carry ancestral material to the right of the current location. Hence, we only need to record the location and the time of the second part of the gene flux events for each lineage.
We note that the coalescent times will not be finite at the breakpoints x=0 and x=1 if the inversion is assumed infinitely old. One can, however, pick two points x0 and x1 arbitrarily close to 0 or 1, respectively, and simulate the interval [x0, x1]. Picking x0 and x1 is step 1 in the algorithm (see below). The process is then initialized by construction of a coalescent tree at x=x0. The next step is to calculate the distance along the chromosome to the next homokaryotypic recombination event (step 3). This distance, Δx, is exponentially distributed with parameter

where LI(x) or LS(x) denotes the total length of the subtree of inverted or standard lineages respectively. If a gene flux event with β2∈(x, x+Δx) occurred previously (for example, during the construction of the coalescence tree at position x), the homokaryotypic recombination event is discarded and the current position is set to x=β2 and Δx=β2−x (step 4). Otherwise the current location is incremented by Δx (step 5).
The final step is performing the recombination or gene flux event, which consists of creating a floating lineage and reattaching the lineage to the remaining tree (see Figure 3 for an illustration of these steps). In the case of a gene flux event, the exact position and time of the event is known from the history of the lineage (see Figure 3b, step 4 in the algorithm below). Homokaryotypic recombination can occur in a standard lineage or an inverted lineage (with probabilities proportional to the length of the subtrees) and the location of the event is picked uniformly on the corresponding subtree (see Figure 3a, step 5 in the algorithm). Then, the older part of the branch is erased—including the history of gene flux of the branch (see Figure 3) and the floating lineage is attached to the remaining tree (step 6).

Sketch of the SMC algorithm with inversions for a sample of size 2. We sample one standard and one inverted chromosome. The dotted line separates the two chromosome classes; lineages to the left of the line belong to the standard class, lineages to the right to the inverted class. Each panel shows three steps: locating the recombination event, creating the floating lineage, and reattaching the floating lineage. Panel a shows a homokaryotypic recombination event, indicated by a crossmark, and the left part of a gene flux event, indicated by an open circle. Panel b shows the right part of the same gene flux event, indicated by a filled circle. Note that another gene flux event is necessary for coalescence. Bold lines show the reattached lineages.
Putting all this together, we can summarize the algorithm in the following steps:
-
1
Pick a starting point x0 and an ending point x1, with 0<x0<x1<1.
-
2
Construct the initial coalescent tree at x0. Record the point in time and the positions on the chromosome of gene flux events for each lineage.
-
3
Draw the distance to the next homokaryotypic recombination from an exponential distribution with parameter
 .
. -
4
Check if we need to update the current tree, that is, if a gene flux event with β2<x+Δx occurred previously somewhere on the tree. If so, update the tree by moving the lineage in which the gene flux event occurred in the other chromosome class, and reattaching it to the tree. Then go to step 3. If there was no gene flux event with β2<x+Δx, continue at 5. If x+Δx>x1, stop.
-
5
Pick a point on the tree for the next homokaryotypic recombination event as described above.
-
6
Erase the older part of this branch (up to the next coalescent event), including its history of gene flux events, that is, all gene flux events that occurred in this lineage. Reattach the lineage to the remaining tree as described above. Go to step 3.
 .
.A detailed illustration of the algorithm can be found in Figure A1 in Appendix A. We note that the original sequential coalescent algorithm requires that the marginal distribution of genealogies is independent of the location on the chromosome. Usually, in inversions, the rate of gene flux between different arrangements varies along the chromosome. As only lineages that belong to the same chromosome class are allowed to coalesce, the rate of gene flux between types influences the rate at which lineages coalesce and the marginal distribution of genealogies is no longer independent from the location on the chromosome. This means that our approach introduces a bias in the marginal distribution of coalescence times if the rate of gene flux is varying along the chromosome. This bias should be negligible if recombination in homokaryotypes is sufficiently frequent, that is, if ρ is large. Roughly speaking, if homokaryotypic recombination is frequent, the step-size at which we move along the chromosome will be small and so will be the change in the rate of gene flux in heterokaryotypes. Consequently, the marginal distributions of genealogies at two consecutive recombination points on the chromosome will be very similar.
Results
To test the accuracy of the algorithm, we implemented a program that simulates genealogies of samples of two chromosomes using the SMC algorithm. We compare the simulations of the SMC to exact coalescent simulations of the ARG and to a forward-in-time Wright–Fisher model (see Appendix for details, the source code of the programs used for the simulations is available on request from the authors). We simulated three different scenarios: both sampled chromosomes are standard, both are inverted, or one is standard and one inverted. To compare the results obtained by the different approaches, we calculated the marginal distribution of coalescence times and the correlation between coalescence times at different loci. We simulated 106 replicates for the SMC algorithm and for the ARG, and 104 replicates in the forward-in-time simulations (FTS). The ARG and the FTS were simulated for a set of five loci (located at x=0.01, 0.1, 0.2, 0.25 and 0.9).
We assume that the inversion is maintained at intermediate frequency by some sort of strong selection. As mentioned before, the inversion is assumed infinitely old. This assumption simplifies the model and should be a good approximation for inversions much older than Ne generations. Recombination in heterokaryotypes is modeled as double crossing-over. The distance between the two cross-over events is fixed at 0.3. Furthermore, the left (right) crossing-over points are uniformly distributed in the interval [0, 0.7] ([0.3, 1]). Then, the rate of gene flux is constant in [0.3, 0.7], and it decreases linearly to zero as we approach the breakpoints. This yields that

The main focus is on a population of size N=500, the largest size that was computationally feasible for our forward-time simulation. We set the map length of the inversion to r=0.1 and the gene flux coefficient to c=10−2. Gene flux per generation and per site then varies from 0 at the breakpoints to ≈0.4 × 10−4 in the interior of the inversion. In addition, we also performed simulations of the SMC with N=50 000. In these simulations, the gene flux coefficient was set to c=10−4 such that the rate of gene flux per site and generation is the same as with N=500 and c=10−2. As extensive simulations of the FTS and the ARG are unfeasible in this case, we compare the outcome of the SMC to analytical predictions for expected coalescence times (Guerrero et al., 2012).
Coalescence times
The distributions of coalescence times at individual sites are a major focus of interest. The reason is that the expected amount of neutral polymorphism is proportional to the coalescence time under many biologically relevant conditions (Wakeley, 2008). Figures 4a and c compare the distributions of coalescence times for a sample of two standard chromosomes. (The results for two inverted chromosomes are very similar and not shown.) Figures 4b and d compare these distributions for a sample of one standard and one inverted chromosome. Figures 4a and b show the distribution of coalescence times for three sites (x=0.01, 0.1, 0.25). As expected, the marginal distribution of coalescent times in the SMC is not exactly the same as in the ARG. In both cases, however, the differences between the SMC and the ARG are very small. In general, the FTS lead to larger coalescence times compared with the ARG and SMC. This is expected because of the small population size N=500 (cf. Equation 3.14 and Figure 3.2 in Wakeley, 2008). Figures 4c and d show the median, and lower and upper quartile of the distributions for better comparison. Median and quartiles fit very well in all cases. Figures 4e and f compare the expected time to coalescence for different population sizes with analytical results (Guerrero et al., 2012) and show that the accuracy of the algorithm increases with increasing N.

Comparison of the distribution of coalescence times. (a, b) Distribution of coalescence times for samples of size 2 at sites x=0.01, 0.1 and 0.25. (c, d) Statistics that summarize the distributions. The boxes show the median, and the lower and upper quartiles of the distribution of coalescence times at sites x=0.01, 0.1, 0.2, 0.25 and 0.9. The whiskers show the minimum and the maximum of the coalescence times. Population size is N=500 and c=10−2 in panels (a, d). (e and f) Expected coalescence time for different population sizes. Circles and crossmarks correspond to results from simulation of the SMC for N=500 and N=50 000, respectively. Solid gray line shows the analytical prediction for expected coalescence time.
Correlation of coalescence times
The correlation of coalescence times at a pairs of site is another quantity important to understanding patterns of neutral genetic variation because it is closely related to measures of LD (McVean, 2002; Wakeley, 2008). The SMC algorithm allows us to visualize correlation of coalescence times for pairs of sites within the whole inverted region for the first time. Figure 5 shows a heat map of these correlation coefficients. Figure 5 illustrates that in the presence of an inversion polymorphism, correlation between sites depends not only on the distance between the two sites but also on their location in the inversion. Correlation of coalescence times is lowest for pairs of sites located at some distance from each other in the interior of the inversion (white islands in Figure 5). In contrast, if one of the sites is located close to one of the breakpoints, coalescence times are more strongly correlated. Another difference to models without inversions is that correlation in coalescence time does not vanish as the distance between sites increases. In fact, correlation may even increase with increasing distance if one site is located close to one of the breakpoints (see local peaks located at coordinates (1, 0) and (0, 1) in Figure 5). The asymmetry in panel Figure 5a is due to the small population size of N=500 and vanishes if N increases (see panel B).

Correlation between coalescent times. Contour plot that illustrates the correlation of coalescence times for pairs of sites if chromosomes are sampled randomly from the population. Dark regions correspond to high levels of correlation and light regions correspond to low levels of correlation. Population sizes are N=500 (a) and N=50 000 (b).
Figures 6a and b show comparisons of the results obtained by the different approaches for N=500. The patterns of correlation generated by the SMC algorithm are reasonably accurate. Again, the results obtained by the SMC are intermediate between the results obtained by the ARG and the FTS. In general, correlation seems to be slightly higher in the SMC than in the ARG. This is in contrast to other versions of the SMC algorithm in which correlation decreases slightly faster than in the ARG. However, we did not simulate sites located very close to each other in the ARG. For such sites, the correlation might indeed decay faster in the SMC with inversions than in the ARG.

Correlation of coalescence times between pairs of sites. The location of the first site is fixed at x=0.1 (a) or x=0.9 (b), and the location of the second site is given on the x-axis. Solid lines correspond to the SMC algorithm, crossmarks correspond to the ARG, and circles to the FTS.
Performance
The main motivation to develop the SMC algorithm for inversions is because it is faster than simulation of the ARG and it requires much less memory. Here, we compare the performance of the different algorithms. Table 1 compares the runtime of the SMC with an implementation of the ARG. All parameter values are chosen as described above. Simulations were done on a single CPU with 8 GB memory. As expected, the SMC algorithm is much faster than the ARG. A sample of two standard chromosomes is roughly 30-times faster in the SMC, and a sample of one standard and one inverted is roughly 15-times faster in the SMC. Note that we simulated only five sites in the ARG (see above) whereas the whole inverted region is simulated in the SMC. Simulation of the ARG for 10 or more sites usually required >8 GB of memory and could not be completed. The differences in performance will be even larger if the size of the inversion, r, or population size, N, increases (McVean and Cardin, 2005).
Discussion
In this paper, we extended the sequential Markovian coalescent to non-standard patterns of recombination induced by inversion polymorphism. One way to think about inversions is by analogy to a model of two populations connected by migration (see Kirkpatrick, 2010). In this analogy, the different chromosome arrangements act as populations and gene flux acts as migration between standard and inverted chromosomes. Based on these similarities, we proposed to simulate gene flux between chromosome arrangements while going backwards in time, and homokaryotypic recombination while moving along the chromosome. This is similar to the SMC with spatial structure (Chen et al., 2009; Eriksson et al., 2009). However, there are important differences. First, it is only part of the chromosome that ‘migrates’. Second, the rate of ‘migration’ between contexts varies within the inverted region. This introduces additional complexity to the ancestral process that describe the genealogical history of samples.
The algorithm allowed us to visualize patterns of correlation of coalescence times between pairs of sites over an entire inverted region for the first time (see Figure 5). As correlation of coalescence times is tightly linked to measures of LD (McVean, 2002), this yields new insights into patterns of LD within inverted regions. Our results show that these patterns differ qualitatively from patterns of LD in standard models without inversions. First, inversions maintain long-distance associations between pairs of sites (Figures 5 and 6). This is similar to models of spatial structure (Ohta, 1982; Wakeley and Lessard, 2003). To understand this, it is helpful to recall our analogy to models of spatial structure. Population structure builds up statistical associations between sites while migration/gene flux erodes it, leading to non-zero values of LD at equilibrium. The analogy is not complete insofar as gene flux varies within the inverted region and sites located close to breakpoints have a lower chance to ‘migrate’ between chromosome classes than sites in the interior of the inversion. As a consequence, in inversions, LD can increase with increasing distance between sites (see Figures 5 and 6). This also means that the amount of LD between two loci depends not only on the distance at which they are located from each other, but also on their location in the inversion.
We compared the results obtained by our algorithm with results obtained from the ancestral recombination graph and a forward-in-time implementation of a Wright–Fisher model. In particular, we compared the marginal distribution of coalescence times, the correlation of coalescence times for pairs of sites, and the speed of the simulations. We found that the marginal distribution of coalescence times is approximated very well by the SMC with inversions (see Figure 4). Comparison with the ARG and the FTS showed that the simulated patterns of correlation are quite accurate (see Figure 6). Remarkably, long-distance correlation is approximated very accurately by the SMC. Table 1 shows a comparison of the run time of the SMC and the ARG. Simulation of 106 replicates of a sample of two inverted (or two standard) chromosomes took about 6 min on a single processor. In contrast, simulation of five sites with the ARG took more than 3 h. To compare these results, it is important to observe that the SMC gives us the gene trees for all sites, whereas the ARG was simulated for a set of five loci. We also tried to simulate more sites with the ARG; simulation of 10 sites within the inversion often required more than 8 GB of memory.
The original SMC, derived as an approximation to the ARG, recovers the exact distribution of marginal coalescent times. This follows from the fact that the distribution of coalescence times is independent of the location on the chromosome (see also Wiuf and Hein, 1999). In inversions, this is usually not the case (Guerrero et al., 2012), which leads to a bias in the marginal distribution of coalescence times in the SMC with inversions. Our results show, however, that this bias is relatively small if N is small and decreases with increasing N (see Figures 4e and f). This is sensible because ρ increases with N and hence the step-size at which we move along the chromosome becomes smaller. Consequently, the distribution of coalescence times does not change much between two consecutive steps. In sum, we expect the SMC algorithm to perform very well whenever population size (N) and/or the inversion (r) is large (see Figures 4e and f, and Figure 5b). Importantly, these are exactly the conditions under which simulation of the ARG becomes unfeasible.
As is common in coalescent approaches, we here assumed that individuals mate randomly. In many systems that are polymorphic for an inversion, however, individuals with the same karyotype tend to mate with each other more often than with individuals with different karyotypes (Kirkpatrick, 2010). The main effect of positive assortment with respect to karyotype class is that the frequency of heterokaryotypes will be reduced relative to their frequency under random mating. Going backwards in time, this means that the probability that a lineage finds itself in a heterokaryotype will be reduced compared with random mating. Thus, flux between chromosome classes will be reduced and neutral divergence between chromosome classes should be larger than expected under random mating. In our algorithm, one can account for nonrandom mating among karyotypes by using the appropriate probability that a lineage finds itself in a heterokaryotype (see equation (1)).
A crucial assumption of our algorithm is that sites within the inversion are neutral. Selection on sites within inversions can be an important factor in the maintenance of polymorphic inversions (Dobzhansky, 1970; Kirkpatrick and Barton, 2006; Joron et al., 2011). As shown by Guerrero et al., 2012, such situations can create peaks of divergence between chromosome classes that center at the selected sites. We expect that coalescence times between selected sites are highly correlated (that is, they are in strong LD). While it would be very interesting to develop a simulation framework for such cases, we do not see how the SMC framework could be modified to allow for selected sites within the inversion.
In contrast to Guerrero et al. (2012), who focused on inversions maintained polymorphic by migration–selection balance in a two-deme model, we here studied a model of an inversion polymorphism in a single population. It would be very interesting to extend our algorithm to more complicated demographic or spatial scenarios (Chen et al., 2009; Eriksson et al., 2009; Excoffier and Foll, 2011). Bottlenecks or changing population sizes can be viewed as changes in the time-scale of the coalescent process (for example, Nordborg, 2008; Wakeley, 2008). Thus, we believe that an extension of our algorithm should yield accurate results if extended to such scenarios. It is more difficult to predict how the introduction of spatial structure might affect the accuracy of the SMC with inversions. In principle, the algorithm can be improved by keeping track of the last k trees, instead of just the previous one (Chen et al., 2009). Obviously, this improves the accuracy of the algorithm, but is costly in terms of speed and memory.
In a recent study, Cheng et al. (2011) exploited a large inversion in the mosquito Anopheles gambiae to identify locally adapted genes. Based on pooled sequence data, they measured divergence in a sliding window along the inverted region. In agreement with theoretical work (Navarro et al., 1997; Andolfatto et al., 2001; Guerrero et al., 2012), they found that divergence is largest close to the break points and decreases to lower levels in the interior of the inversion. The pattern, however, shows a considerable amount of variation. As suggested by Cheng et al. (2011), the variation may result from spatially varying selection on sites within the inverted region. Potential targets of selection were then identified using an outlier approach. While this approach can indeed successfully identify regions under selection, it is also expected to miss many of the selected sites (Teshima et al., 2006). Further, this interpretation of outliers needs to be treated with caution. Substantial variation in FST can result from the basic stochastic nature of the coalescent process (Guerrero et al., 2012).
These limitations highlight potential applications for our SMC algorithm. It can be used to predict distributions of polymorphism in an inverted region under hypotheses of interest. Importantly, the SMC algorithm also predicts patterns of LD, which harbor information that cannot be obtained from analyzing markers independently (Lawson et al., 2012). The data can then be tested against those predictions. The SMC algorithm can be used to produce neutral null distributions as well as alternatives invoking selection. By simply changing the frequency of the inversion through time, one can simulate neutral inversions or selective sweeps of inversions (Prezeworski et al., 2005; Guerrero et al., 2012). Alternatively, the parameters of a model can be fit to the data using simulations with the SMC method, for example via approximate Bayesian computation (Beaumont, 2010). The ABC framework could also be used to compare different hypotheses and choose the model that fits the data best (Csillery et al., 2010).
In summary, we showed how the SMC algorithm can be extended to simulate neutral genetic variation in inversions. Although the presence of inversions at intermediate frequency introduces bias in the marginal distribution of coalescence times, the accuracy of the extended algorithm is very good. The algorithm is much faster and, importantly, more memory-efficient than the ARG. Hence, this algorithm could be a useful tool to study patterns of genetic variation in inversions in more detail in cases where computation time and memory are limiting factors.
Data Archiving
There were no data to deposit.
References
Andolfatto P, Depaulis F, Navarro A . (2001). Inversion polymorphisms and nucleotide variability in Drosophila. Genet Res 77: 1–8.
Ashburner M . (1989). Drosophila. A laboratory handbook. Cold Spring Harbor Laboratory Press.
Beaumont M . (2010). Approximate Bayesian computation in evolution and ecology. Annu Rev Ecol Evol Syst 41: 379–406.
Bush G, Case S, Wilson A, Patton J . (1977). Rapid speciation and chromosomal evolution in mammals. Proc Natl Acad Sci USA 74: 3942–3946.
Charlesworth D, Charlesworth B, Marais G . (2005). Steps in the evolution of heteromorphic sex chromosomes. Heredity 95: 118–128.
Chen G, Marjoram P, Wall J . (2009). Fast and flexible simulation of DNA sequence data. Genome Res 19: 136–142.
Chen G, Slaten E, Ophoff R, Lange K . (2006). Accommodating chromosome inversions in linkage analysis. Am J Hum Genet 79: 238–251.
Cheng C, White B, Kamdem C, Mockaitis K, Costantini C, Hahn M et al. (2011). Ecological genomics of anopheles gambiae along a latitudinal cline in Cameroon: a population resequencing approach. Genetics 190: 1417–1432.
Chovnick A . (1973). Gene conversion and transfer of genetic information within the inverted region of inversion heterozygotes. Genetics 75: 123–131.
Csillery K, Blum M, Gaggiotti O, Francois O . (2010). Approximate bayesian computation (ABC) in practice. Trends Ecol Evol 25: 410–418.
Dobzhansky T . (1970) Genetics of the Evolutionary Process Vol 139, Columbia University Press.
Eriksson A, Mahjani B, Mehlig B . (2009). Sequential Markov coalescent algorithms for population models with demographic structure. Theoretical Population Biol 76: 84–91.
Ewing G, Hermisson J . (2010). MSMS: A coalescent simulation program including recombination, demographic structure and selection at a single locus. Bioinformatics 26: 2064–2065.
Excoffier L, Foll M . (2011). Fastsimcoal: a continuous-time coalescent simulator of genomic diversity under arbitrarily complex evolutionary scenarios. Bioinformatics 27: 1332–1334.
Griffiths R, Marjoram P . (1996). Ancestral inference from samples of DNA sequences with recombination. J Comput Biol 3: 479–502.
Griffiths R, Marjoram P . (1997). An ancestral recombination graph. Institute for Mathematics and its Applications 87: 257–270.
Guerrero R, Rousset F, Kirkpatrick M . (2012). Coalescent patterns for chromosomal inversions in divergent populations. Phil Trans R Soc B 367: 430–438.
Hoffmann A, Rieseberg L . (2008). Revisiting the impact of inversions in evolution: from population genetic markers to drivers of adaptive shifts and speciation? Annu Rev Ecol Evol, Syst 39: 21–42.
Hudson R . (1983). Properties of a neutral allele model with intragenic recombination. Theoret Population Biol 23: 183–201.
Hudson R . (1990). Gene genealogies and the coalescent process. Oxford Surveys in Evolutionary Biology Vol. 7. Oxford University Press: New York. pp 1–44.
Hudson R . (2002). Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18: 337–338.
Joron M, Frezal L, Jones R, Chamberlain N, Lee S, Haag C et al. (2011). Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477: 203–206.
Kennington WJ, Partridge L, Hoffmann AA . (2006). Patterns of diversity and linkage disequilibrium within the cosmopolitan inversion In(3R)Payne in Drosophila melanogaster are indicative of coadaptation. Genetics 172: 1655–1663.
Kirkpatrick M . (2010). How and why chromosome inversions evolve. PLoS Biol 8: e1000501.
Kirkpatrick M, Barton N . (2006). Chromosome inversions, local adaptation and speciation. Genetics 173: 419–434.
Kirkpatrick M, Kern A . (2012). Where’s the money? Inversions, genes, and the hunt for genomic targets of selection. Genetics 190: 1153–1155.
Laayouni H, Hasson E, Santos M, Fontdevila A . (2003). The evolutionary history of Drosophila buzzatii. XXXV. Inversion polymorphism and nucleotide variability in different regions of the second chromosome. Mol Biol Evol 20: 931–944.
Laval G, Excoffier L . (2004). Simcoal 2.0: a program to simulate genomic diversity over large recombining regions in a subdivided population with a complex history. Bioinformatics 20: 2485–2487.
Lawson D, Hellenthal G, Myers S, Falush D . (2012). Inference of population structure using dense haplotype data. PLoS Genet 8: e1002453.
Marjoram P, Wall J . (2006). Fast “coalescent” simulation. BMC Genetics 7: 16.
McGaugh S, Noor M . (2012). Genomic impacts of chromosomal inversions in parapatric drosophila species. Phil Trans R Soc B 367: 422–429.
McVean G . (2002). A genealogical interpretation of linkage disequilibrium. Genetics 162: 987–991.
McVean G, Cardin N . (2005). Approximating the coalescent with recombination. Phil Trans R Soc B 360: 1387–1393.
Navarro A, Barbadilla A, Ruiz A . (2000). Effect of inversion polymorphism on the neutral nucleotide variability of linked chromosomal regions in drosophila. Genetics 155: 685–698.
Navarro A, Barton N . (2003). Accumulating postzygotic isolation genes in parapatry: a new twist on chromosomal speciation. Evolution 57: 447–459.
Navarro A, Betran E, Barbadilla A, Ruiz A . (1997). Recombination and gene flux caused by gene conversion and crossing over in inversion heterokaryotypes. Genetics 146: 695–709.
Nordborg M : Coalescent Theory. In: Balding DJ, Bishop M, Cannings C (eds) Handbook of Statistical Genetics 3rd edn John Wiley & Sons, Ltd: Chichester, UK. (2008).
Novitski E, Braver G . (1954). An analysis of crossing over within a heterozygous inversion in Drosophila melanogaster. Genetics 39: 197–209.
Ohta T . (1982). Linkage disequilibrium with the island model. Genetics 101: 139–155.
O’Reilly P, Coin L, Hoggart C . (2010). invertFREGENE: software for simulating inversions in population genetic data. Bioinformatics 26: 838–840.
Prezeworski M, Coop G, Wall J . (2005). The signature of positive selection on standing genetic variation. Evolution 59: 2312–2323.
Ranz J, Maurin D, Chan Y, Von Grotthuss M, Hillier L, Roote J et al. (2007). Principles of genome evolution in the Drosophila melanogaster species group. PLoS Biol 5: e152.
Roberts P . (1976). The genetics of chromosome aberration. The Genetics and Biology of Drosophila. Academic Press: London, UK, 67–184.
Teshima KM, Coop G, Przeworski M . (2006). How reliable are empirical genomic scans for selective sweeps? Genome Res 16: 702–712.
Wakeley J . (2008) Coalescent Theory. CSIRO: USA.
Wakeley J, Lessard S. . (2003). Theory of the effects of population structure and sampling on patterns of linkage disequilibrium applied to genomic data from humans. Genetics 164: 1043–1053.
White M . (1978). Chain processes in chromosomal speciation. Syst Biol 27: 285–298.
Wiuf C, Hein J . (1999). Recombination as a point process along sequences. Theoret Population Biol 3: 248–259.
Acknowledgements
This work was funded by NSF grant DEB-0819901 to MK. We thank three anonymous reviewers for helpful comments on an earlier version of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Appendix
Appendix
(A) Example of the SMC with inversions
We here describe an example of the SMC with inversions to illustrate the algorithm in more detail. Figure A1 shows an illustration of the steps performed in the algorithm. The following steps are shown in Figure A1:
-
1
The process is initiated by constructing a coalescent tree at the initial position x0 (indicated by the arrow). A gene flux event F1 in chromosome 1 moves the ancestral material bounded by f1 and f1′ from a standard into an inverted chromosome. This allows the two lineages to coalesce.
-
2
The current position is incremented to the position of the next homokaryotypic recombination event H1, which occurs in chromosome 2 at position h1. The vertical line indicating the left boundary of the gene flux event F1 is removed from chromosome 1 because it is to the left of the current position. The branch above H1 is removed and a new gene tree is created.
-
3
The current point is incremented to the position of the next homokaryotypic recombination event H2. Gene flux events change the context of the ancestral material from chromosome 2 (F2) and chromosome 1 (F3) before the lineages coalesce.
-
4
The next homokaryotypic recombination event is further to the right than the right-hand boundary of the gene flux event F1. Thus, the gene flux event occurs instead of a homokaryotypic recombination event. Note that the right part of the gene flux event F3 is removed in this step. This is because the ancestral material to the right of the current position recombined into a different genetic background before the gene flux event F3 occurred.
-
5
The current position is incremented to the position of the next homokaryotypic recombination event H3.
-
6
The next homokaryotypic recombination event is further to the right than the right-hand boundary of the gene flux event F2. Thus, the gene flux event occurs instead of a homokaryotypic recombination event. The two lineages coalesce after gene flux event F4 moves the ancestral material from chromosome 1 into an inverted chromosome.
-
7
The current position is incremented to the position of the next homokaryotypic recombination event H4. Gene flux event F4 is removed because the ancestral material to the right of the current position is not affected by it. The two lineages coalesce after gene flux event F5 moves the ancestral material of chromosome 2 into a standard chromosome.
-
8
After homokaryotypic recombination event H5, the final coalescent tree is constructed.

Cartoon example of the SMC algorithm with inversions for a sample of one standard (Chr 1) and one inverted chromosome (Chr 2). The top part of each panel shows a gene tree, with the thin vertical line separating standard and inverted chromosomes. The bottom part of each panel shows the chromosomes, where the arrow indicates the point to which the tree pertains. Crossmarks and circles indicate the breakpoints of recombination and gene flux events, respectively. The ith gene flux event is labeled Fi and the ith homokaryotypic recombination event is labeled Hi. The boundaries of gene flux events are indicated by dotted vertical lines on the chromosomes, labeled by fi (left boundary) and fi′ (right boundary). The chromosomal position of the ith homokaryotypic recombination event is indicated by a solid vertical line and labeled by hi. Dashed lines in the coalescent tree show the removed part of the tree, that is, the branch above a recombination or gene flux event. The segment of the chromosome for which trees have already been constructed is shaded gray.
(B) Ancestral recombination graph (ARG) with chromosomal inversion polymorphism
We simulate the genealogy of a sample of k chromosomes (carriers) from a population of N diploid individuals going backwards in time. Carriers have ancestral genetic content at sites that we map in terms of recombination distance (r). The population is subdivided in two classes (inverted and standard). Three types of events can occur: coalescence, recombination, and gene flux. The rates of these events are described in the main text (equation (2), see below for details). We add all rates to obtain a single waiting time until the next event, and go to that generation. We then chose one event to execute, based on the relative rates of the three possibilities. After carrying out the event, we update the rate values based on the new number of carriers. We repeat the process until there is one carrier left.
For coalescent events, we randomly choose one of the chromosome classes, weighted by their rates of coalescence (as defined in equation (2), main text). From this class, we draw two random carriers and merge them into one. This process involves combining the ancestral genetic content both carriers have. As a result, the new number of carriers is k−1.
For recombination events, we only consider those between the leftmost and rightmost site held by a carrier at a given time. Therefore, each carrier i has a probability of recombination ri (the distance between its leftmost and rightmost site), and a recombination rate Ri=piri, where pi is the frequency of the chromosome class of carrier i. The population-wide recombination rate is 2N∑Ri, performing the sum of Ri over all carriers. We choose a random carrier, weighting each by Ri, and carry out the recombination event on it. This involves splitting the carrier’s ancestral content in two at a random point (uniform between the leftmost and rightmost sites). We create two ancestral carriers with the left and right halves produced by the split. We assume that there is no recombination between carriers. Thus, recombination always increases k by 1.
The rates of gene flux are as described in equation (2) (main text), with ϕ(x)=1. We choose a random carrier and the boundaries for the gene exchange (β, as in text). We take all ancestral material that falls within the boundaries and assign it to a new carrier (of the opposite chromosome class to the carrier being operated on). Again, because we assume that there is no gene flux between carriers, such event always increases k by 1.
(C) Implementation of the forward-in-time Wright–Fisher model
We implemented a standard forward-in-time Wright–Fisher model for a population of N=500 diploid individuals. Simulations were run for 1000 generations without selection before the inversion was added. Adding the inversion was done by randomly sampling half the chromosomes in the population to be inverted.
We modeled selection as frequency-independent balancing selection. Subsequent generations were created by a weighted sampling of 2N individuals where each sampled individual produced one gamete. In the sampling step, heterokaryotypes were favored over homokaryotypes. The selection coefficient against homokaryotypes was 0.1.
Each generation, recombination occurred with probability r in homokaryotypes. If so, a break point between 0 and 1 was chosen randomly and a gamete was produced accordingly. In heterokaryotypes, gene flux occurred with probability cr. When this happened, a window of length 0.3 was placed randomly within the inverted region of the chromosome and a gamete was produced as described in the main text.
Each simulation was then allowed to progress for 600 N generations, after which three pairs of chromosomes were sampled (standard–standard, inverted–inverted and standard–inverted). For each pair, all five loci were checked for coalescence and the age of their most recent common ancestor was recorded. If all sites had not coalesced, the simulation was considered a failure and the run was disregarded. In total only ∼10% of all simulations failed.
Rights and permissions
About this article
Cite this article
Peischl, S., Koch, E., Guerrero, R. et al. A sequential coalescent algorithm for chromosomal inversions. Heredity 111, 200–209 (2013). https://doi.org/10.1038/hdy.2013.38
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2013.38
Keywords
This article is cited by
-
Advances in Computer Simulation of Genome Evolution: Toward More Realistic Evolutionary Genomics Analysis by Approximate Bayesian Computation
Journal of Molecular Evolution (2015)
