
Abstract
In the last few years, dozens of studies have documented the detection of loci influenced by selection from genome scans in a wide range of non-model species. Many of those studies used amplified fragment length polymorphism (AFLP) markers, which became popular for being easily applicable to any organism. However, because they are anonymous markers, AFLPs impose many challenges for their isolation and identification. Most recent AFLP genome scans used capillary electrophoresis (CE), which adds even more obstacles to the isolation of bands with a specific size for sequencing. These caveats might explain the extremely low number of studies that moved from the detection of outlier AFLP markers to their actual isolation and characterization. We document our efforts to characterize a set of outlier AFLP markers from a previous genome scan with CE in ocellated lizards (Lacerta lepida). Seven outliers were successfully isolated, cloned and sequenced. Their sequences are noncoding and show internal indels or polymorphic repetitive elements (microsatellites). Three outliers were converted into codominant markers by using specific internal primers to sequence and screen population variability from undigested DNA. Amplification in closely related lizard species was also achieved, revealing remarkable interspecific conservation in outlier loci sequences. We stress the importance of following up AFLP genome scans to validate selection signatures of outlier loci, but also report the main challenges and pitfalls that may be faced during the process.
Similar content being viewed by others

Introduction
The use of population genomic approaches to search for genomic regions potentially under selection has gained much popularity in the last decade. Amplified fragment length polymorphism (AFLP) markers have been used frequently for genome scans in several non-model species (for example, Bonin et al., 2006; Minder and Widmer, 2008; Apple et al., 2010). First described by Vos et al. (1995), the AFLP technique consists of the digestion of genomic DNA with restriction enzymes, the ligation of adaptors to the digested fragments and the amplification by PCR of these fragments using selective primers that anchor in the adaptors. Hundreds or even thousands of polymorphic AFLP markers, distributed across the whole genome, can be easily and affordably genotyped for many populations of any species, but they are scored as dominant markers and the sequence content of each AFLP marker remains unknown throughout the whole process. Although new developments in next-generation sequencing technologies are offering affordable ways to scan the genome for loci under selection and overcome marker anonymity (Rowe et al., 2011), there are still a large number of AFLP-based projects in which it is highly worthwhile to determine the sequence content of outlier loci suspected to be under selection.
Recent efforts have been made to improve the reliability of existing methods for statistical detection of outlier loci in AFLP genome scans, by controlling factors that inflate the false-positive rate, such as homoplasy (that is, comigration of non-homologous bands with the same size), population structure and history or multiple comparisons (Caballero et al., 2008; Excoffier et al., 2009; Pérez-Figueroa et al., 2010). However, the profusion of methodologies employed among genome-scan studies to detect outliers and the use of variable criteria for outlier classification (use of one or several methods simultaneously; variable significance thresholds; population pairwise comparisons versus global analyses) makes it difficult to compare results from different taxa (Butlin, 2010). We could learn much more from such studies if AFLP markers detected as outliers were brought out from anonymity and further research were conducted toward the identification of genes linked with such outliers and their implications for local adaptation. Unfortunately, and despite >25 AFLP genome scans for selection in non-model species available in the literature, only a few studies report their attempts to isolate and sequence AFLP markers identified as candidate loci under selection (Minder and Widmer, 2008; Wood et al., 2008, Midamegbe et al., 2011; Paris and Despres, 2012). The isolation of a particular AFLP fragment can be technically demanding and time consuming, often involving the need for fragment cloning. The use of capillary electrophoresis (CE) has become popular for separating fluorescently labelled AFLP fragments, with gains in both resolution and sensitivity as compared with traditional polyacrylamide gels with silver staining (Polanco et al., 2005; Apple et al., 2010). Nevertheless, CE adds extra difficulties to the isolation of AFLP fragments because they can no longer be directly excised from the denaturing matrix. These technical drawbacks may help to explain why most AFLP markers identified as candidate loci potentially under selection in genome scans still remain completely anonymous.
Despite the troublesome nature of the AFLP marker's isolation and identification, the follow-up of AFLP genome scans can provide new candidate genes or regulatory elements with importance in adaptation that were unsuspected before. That was the case reported in Wood et al. (2008), who found no differentiation in flanking regions of sequenced outliers, indicating that indel polymorphisms detected within outlier sequences (with characteristics of transposable elements) could be the actual targets of selection, perhaps affecting the expression of downstream loci.
The analysis of outlier AFLP sequences is necessary to determine the sources of length polymorphism that are responsible for the outlier behaviour. Although base substitutions at enzyme restriction sites or at selective bases are normally described as the main cause of fragment length polymorphism (Bensch and Akesson, 2005), other mutations such as insertions or deletions (indels) or polymorphic microsatellites occurring between restriction sites have also been reported as sources of length polymorphism (Meksem et al., 2001; Wong et al., 2001; Wood et al., 2008). Although sequences from visible AFLP alleles (dominant alleles) are expected to be conserved among populations, the opposite is true for recessive alleles, because any fragment different enough to migrate faster or slower than the dominant allele will be scored as absent. Thus, the conversion of outlier AFLP markers into codominant markers through the design of specific primers offers the chance to investigate sequence variation both in dominant and recessive alleles.
Here we report the results and main difficulties faced when trying to isolate and characterize a set of outliers resulting from a previous AFLP genome scan conducted by Nunes et al. (2011a) in the European ocellated lizard (Lacerta lepida). The species is widespread throughout the Iberian Peninsula in a variety of ecological conditions that are strongly influenced by the distribution of precipitation and temperature ranges. Morphological and genetic differentiation in populations from the northwest and southeast of the Iberian Peninsula are strong enough to consider those populations as distinct subspecies: Lepida lepida iberica and Lepida lepida nevadensis, respectively (Mateo and Castroviejo, 1990; Paulo et al., 2008). The first lives in a rainy and less-warm weather regime, whereas the second inhabits a region that experiences hot summers and the lowest annual rainfall across the species’ distribution range. Detection of selection with DFDIST (Beaumont and Nichols, 1996) and BayeScan (Foll and Gaggiotti, 2008) produced a combined list of 23 AFLP outliers (5.9% of investigated loci) targeted for further validation. Nunes et al. (2011a) also tested for associations between AFLP band frequency and variation in climatic variables across the Iberian Peninsula with the spatial analysis method (Joost et al., 2008). Several loci detected as outliers were also associated with temperature, insolation or precipitation.
Materials and methods
Isolation and cloning of outlier AFLP markers
Twelve AFLP markers from a list of 23 outliers considered by Nunes et al. (2011a) as candidate loci potentially under selection were chosen for isolation (Table 1). AFLP outliers were detected through global analyses with 10 genotyped populations: Galicia (GAL) and Gerês (GER) from L. l. iberica subspecies; Béjar (BEJ), Serra da Estrela (SET), Peniche (SPE), Castro Marim (CMA), Alentejo (ALE), Toledo (TOL) and Andalucia (AND) from L. l. lepida; Almeria (ALM) from L. l. nevadensis (see Nunes et al., 2011a for further details). The set of 12 outliers selected for isolation consisted of the five outliers detected by both DFDIST and Bayescan (markers 245, 311, 315, 386 and 390), four randomly chosen outliers among the ones detected by DFDIST only (markers 140, 209, 297 and 301) and another three outliers among the ones detected by BayeScan only (markers 75, 235 and 323). The first step in the isolation of outliers consisted of the re-amplification of 2–4 samples for each outlier, where the band was scored as present. PCR reactions were conducted with digested DNA and the same conditions as in Nunes et al. (2011a), but using an EcoRI selective primer without fluorescent label (to avoid interference in downstream steps) and a Green GoTaq Flexi PCR buffer (Promega, Madison, WI, USA) for direct loading of PCR products into agarose gels. For each sample, three PCR replicates (10 μl × 3) were loaded together in the same lane of a 1.5% agarose gel stained with ethidium bromide. Three contiguous slices of gel were excised from each lane within a size range of 50–100 bp that included the desired outlier size (Figure 1). Each gel slice was purified separately with GENECLEANII kit (MP Biomedicals, Santa Ana, CA, USA) to recover the DNA fragments. To confirm the recovery of the desired AFLP marker, gel purified fragments were used as template for a PCR with fluorescent labelled primers as in Nunes et al. (2011a). PCR products were then separated by CE on an ABI Prism 310 (Applied Biosystems, Foster City, CA, USA) to verify which gel-slice DNA produced the fragment with the desired size and should be used in the cloning reaction. The TOPO TA Cloning Kit (Invitrogen, Paisley, UK) was used for outlier fragment cloning according to the manufacturer’s instructions. DNA isolated from the gel was amplified with unlabelled primers using the original PCR conditions reported in Nunes et al. (2011a), and then cloned through direct insertion into a plasmid vector and transformed into Escherichia coli cells. Single colonies were randomly selected to construct libraries with 96–192 clones for each outlier AFLP marker.

Schematic representation of the steps used to isolate and sequence outlier AFLP marker 75, with a band size of 193 base pairs. A full color version of this figure is available at the Heredity journal online.
Library screening
Because each cloning reaction was expected to include multiple fragments of similar size to the outlier fragment, the following procedure was used to identify clones bearing inserts with the desired size before sequencing, thus reducing the number of clones to be sequenced (Figure 1). First, each colony from a library was amplified by PCR with universal primers M13 in a total reaction volume of 15 μl. The amplified clones were readily used as template for another PCR with fluorescent-labelled selective primers using the same conditions as Nunes et al. (2011a), but scaled to a final volume of 5 μl. The size of this PCR product will be the one of the DNA fragment inserted into the plasmid, and can be used to determine if the clone insert has the expected size and should be sequenced. The size of inserts in each clone was determined by CE, with PCR products pooled in sets of 12. If fragments with the expected size were present within a pool of inserts, the respective clones were run separately to identify which of the 12 clones was bearing the insert of the expected size (Figure 1). Only 1–3 clones with inserts of the desired size (confirmed by CE) were sequenced for each outlier, with M13 primers and using standard protocols (BigDye Terminator v.3.1, Applied Biosystems) on an ABI PRISM 310 (Applied Biosystems). Sequences were edited in Sequencher v.4.0.5 (Gene Codes Co., Ann Arbor, MI, USA) and deposited in GenBank.
Outlier sequence characterization
Cloned sequences were aligned with sequences of EcoRI and MseI selective primers to check for mismatches in selective bases. GenBank was searched for sequences homologous to each clone insert sequence using BLASTN. Sequences were also inspected for the presence of open-reading frames that could indicate that the sequence might correspond totally or partially to a coding region.
An internal primer pair for each sequenced outlier was designed as close to the sequence ends as possible using Primer 3 (Rozen and Skaletsky, 2000). The Reddy et al. (2008) method for genome-walking was employed to extend outlier fragment sequences into their flanking regions, but all attempts failed. To investigate sources of polymorphism between the dominant allele (scored as present) and the recessive alleles (scored as absent), we combined unlabelled EcoRI or MseI selective primer with the complementary outlier-specific primer in two independent amplifications to obtain the full sequence from recessive alleles. For each outlier, digested DNA from one sample where the outlier was scored as absent (homozygous for the recessive allele) was used for PCR with 1x PCR buffer (Promega), 0.75 U GoTaq DNA polymerase (Promega), 2.0 mM MgCl2, 0.12 mM dNTPs and 0.4 μM of each primer in a final volume of 15 μl. The cycling conditions used were 3 min at 94 °C, 35 × (30 s at 94 °C, 30 s at outlier-specific primer annealing temperature (Table 1), and 30 s at 72 °C), followed by 10 min at 72 °C. Purified products (Sureclean, Bioline, London, UK) were sequenced in both directions using standard protocols (BigDye Terminator v.3.1, Applied Biosystems) on an ABI PRISM 310 (Applied Biosystems).
Internal primer pairs designed for each outlier were tested in undigested DNA from Lacerta lepida samples, previously genotyped by Nunes et al. (2011a), to evaluate primer efficiency in the conversion of outlier AFLPs into codominant markers. For those outliers whose internal primers worked properly, sequences were obtained for 20–27 out of 196 samples genotyped in the AFLP genome scan (6–9 samples from L. l. iberica, 8–12 samples from L. l. lepida and 6 samples from L. l. nevadensis) to corroborate outlier AFLP scoring and to characterize sequence variation in both dominant and recessive alleles (Supplementary Table S1). The same outlier-specific primers were tested in cross-species amplification in African ocellated lizards (three samples from L. pater and 4–7 samples from L. tangitana), in one Schreiber’s green lizard (L. schreiberi), one Iberian rock lizard (Iberolacerta monticola) and one sand lizard (L. agilis) (Supplementary Table S1). PCR reactions and sequencing were performed as above.
Sequences were edited in Sequencher v.4.0.5 (Gene Codes Co.). Sequences of each allele from samples that were heterozygous in length were reconstructed according to guidelines from Flot et al. (2006). Base ambiguities were resolved with PHASE 2.1.1 (Stephens et al., 2001; Stephens and Scheet, 2005). We ran the algorithm five times (1000 iterations with the default values) with different random-number seeds, and the same haplotypes were consistently recovered in each run. Phased alleles from each individual were aligned with CLUSTAL W (Thompson et al., 1994) as implemented in Bioedit (Hall, 1999), and gap length for repetitive element alignment was adjusted manually. Sequences from haplotypes detected in each lizard species and each outlier AFLP marker were deposited in GenBank. Nucleotide diversity (π) and haplotype diversity (H) for each outlier were determined for each ocellated lizard species and subspecies in ARLEQUIN 3.5 (Excoffier et al., 2005). Neutrality was tested with Tajima’s D test (Tajima, 1989) for each ocellated lizard species or subspecies. To infer the relationships among haplotypes, a minimum spanning network was constructed for each outlier marker with the median-joining method (Bandelt et al., 1999) in NETWORK 4.51 (www.fluxus-engineering.com). The input file was converted from fasta to nexus format with CONCATENATOR 1.1.0 (Pina-Martins and Paulo, 2008).
Results
AFLP marker isolation and sequence
All the 12 outlier AFLP markers were successfully isolated from agarose gel slices, re-amplified and cloned. Clones with the expected size were retrieved for only seven outliers (Table 1). Mismatches in the EcoRI or MseI primer-selective bases were not detected in sequenced clones and their sequences did not align with each other, indicating that the seven outlier AFLPs belong to independent loci. No open-reading frames could be detected in outlier fragment sequences. Their sequences are likely to be non-coding regions and some are quite rich in repetitive elements. Only three outlier sequences returned significant hits when blasted against the GenBank (Table 1), showing some homology with the green anole (Anolis carolinensis) or with the Indian python (Python molurus) whole-genome shotgun sequences, but these species have no known genes annotated around the homologue of the outlier sequence. Because A. carolinensis and P. molurus are distantly related with ocellated lizards, possible inferences on the significance of the homologies detected here are very limited.
A specific primer pair was designed for each outlier based on clone sequences and used to amplify the fragments directly from undigested genomic DNA. The amplification was successful for five loci, mk75, mk209, mk245, mk386 and mk390, but failed for mk140 and mk311. Full-length sequences from recessive alleles were successfully obtained from digested DNA (combining AFLP selective primers with outlier-specific internal primers) for outliers mk209, mk245 and mk386 (Supplementary Figure S1). In mk209, an indel of five bases preceded by three consecutive single-nucleotide polymorphisms explained the length polymorphism, whereas for mk245, the dominant allele carried a microsatellite composed by six GTT repeats, but the recessive allele had only three GTT repeats. For both mk209 and mk245, sources of polymorphism were located within the segment amplified with the internal specific primer pairs. In locus mk386, the amplification of the full-length sequence from the recessive allele revealed a deletion of three bases, just before the binding site of the first specific primer, which probably accounts for the length polymorphism.
All attempts to isolate the full sequence of the mk75-recessive allele failed. Nevertheless, length polymorphism between dominant and recessive alleles of mk75 could be explained by an insertion of nine base pairs in the recessive allele, located within the fragment amplified with mk75 internal primers (Supplementary Figure S1). As for mk390, eight single-nucleotide polymorphisms and a single-base deletion were detected between the dominant and recessive-allele sequences. However, the single-base deletion and three of these single-nucleotide polymorphisms were located in the mk390L1 primer-binding site, preventing the amplification of the MseI adaptor end of the sequence from the recessive allele. Moreover, the use of the mk390 internal primer pair was not successful in converting mk390 into a codominant marker, since only the dominant allele could be amplified (Supplementary Figure S1). For both mk386 and mk390, there were no alternative binding sites to design internal primers suitable to amplify both dominant and recessive alleles from genomic DNA. However, the mk390 locus was amplified and sequenced in African ocellated lizards with the mk390 internal primer pair, which indicates that at least the dominant allele is present and conserved in L. pater and L. tangitana (accession numbers JQ310741-JQ310742).
Intra and interspecific variation in outlier sequences
Outliers mk75, mk209 and mk245 were sequenced from undigested DNA with specific internal primers for 20–27 L. lepida samples, previously genotyped with AFLP markers, by Nunes et al. (2011a) (Table 2). No discordances between band score genotypes and sequences obtained for mk75 were detected. This means that sequences from samples where the band mk75 was scored as absent were carrying two recessive alleles as expected, whereas samples where the band was scored as present had either two dominant alleles or one dominant allele together with a recessive allele. Although only 11 samples with the mk75 band scored as present were sequenced, homozygous individuals for the dominant allele (four samples) were only detected in L. l. iberica populations, whereas sequenced samples from L. l. lepida populations were heterozygous, carrying the expected dominant-allele sequence, but also a recessive allele (Supplementary Table S1). Marker mk75 band frequency recorded in the L. lepida AFLP genome scan increased from southern to north-western populations of the Iberian Peninsula, especially in L. l. iberica populations (Figure 2). The results from mk75 sequences confirmed this trend and the probability for a sample to be homozygous for the dominant allele seemed to be higher in north-west populations.

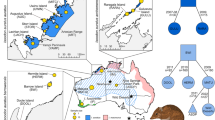
Frequency of band presence observed for outlier AFLP loci mk75, mk209 and mk245 in each Lacerta lepida population genotyped by Nunes et al. (2011a). Populations are displayed according to their geographical position in the Iberian Peninsula, from the northwest to the southeast: Galicia (GAL) and Gerês (GER) from L. l. iberica subspecies; Béjar (BEJ), Serra da Estrela (SET), Peniche (SPE), Castro Marim (CMA), Alentejo (ALE), Toledo (TOL) and Andalucia (AND) from the nominal subspecies, L. l. lepida; Almería (ALM) from L. l. nevadensis subspecies.
A total of 10 mk75 recessive-allele haplotypes were detected in ocellated lizards, differing in single mutations from each other, whereas only a single dominant-allele haplotype could be found, that seems to be derived from a single deletion event of 9 bp (Figure 3a). Amplification of mk75 in African ocellated lizards (L. pater and L. tangitana) revealed the absence of the dominant allele, except for one sample from Morocco that was heterozygous with one dominant allele and the most frequent recessive allele in L. lepida (Figure 3a). The same specific primers were able to amplify the mk75 fragment in L. schreiberi, L. agilis and in Iberolacerta monticola, retrieving remarkably conserved sequences (Supplementary Figure S2).

Minimum spanning haplotype network for the mk75 (a), mk209 (b) and mk245 (c). The size of the circles is proportional to sample size. Each mutation between haplotypes is represented by a dash. Asterisks denote mutations at indel sites. Dominant haplotypes (i.e, the AFLP band scored as present) obtained from European ocellated lizards for each outlier are indicated. Non-ocellated lizard haplotypes are represented in white dashed circles: L. agilis (Lag), Iberolacerta monticola (Imo) and L. shreiberi (Lsc).
Sequencing of 23 L. lepida samples for mk209 resulted in two dominant-allele haplotypes, differing in a single mutation, and found exclusively in L. l. nevadensis samples (Figure 3b, Table 2). Outlier mk209 was scored as present in genome scan genotyping at a high frequency in L. l. nevadensis’ population (ALM=0.94), but was nearly or completely absent in all other populations (<0.1, Figure 2). All samples outside the ALM population for which outlier mk209 was scored as present were sequenced (three samples), but none of them had any copy of a dominant allele, suggesting that homoplasious fragments must have been responsible for the erroneous positive scoring in these three samples (Supplementary Table S1). Five mk209 recessive haplotypes were found in L. lepida and they result mostly from length variation in a repetitive element of Gs that follows the indel TGGA responsible for mk209 length polymorphism (Table 2; Supplementary Figure S3). Sequences from mk209 obtained from African ocellated lizards showed that all investigated samples from L. pater and some from L. tangitana share the insertion of TGGA with the dominant allele, but they are variable in length in the G and GA repetitive elements (Table 2), resulting in 11 recessive haplotypes exclusive to Africa (Supplementary Figure S3). An individual from Morocco, the one carrying European alleles from mk75, also carried an mk209 haplotype that differs in a single mutation from the second most frequent haplotype in European ocellated lizards (Figure 3b). Specific primers designed for mk209 worked well in Iberolacerta monticola, but not in L. schreiberi or L. agilis. The sequence obtained from I. monticola was conserved and homologous to those from ocellated lizards.
Outlier mk245 was sequenced for 20 L. lepida samples and revealed that the microsatellite responsible for length polymorphism was quite variable in L. l. lepida subspecies, with 3–10 GTT repeats (Table 2). Moreover, most L. l. lepida samples analysed (six out of eight samples) were heterozygous for GTT repeat number. The opposite was verified for L. l. iberica and L. l. nevadensis, as all sequenced samples presented haplotypes with three GTT repeats only (Table 2). Nevertheless, haplotype diversity was higher for L. l. nevadensis (H=0.561), whereas in L. l. iberica (H=0.000) all analysed sequences corresponded to the same haplotype (Figure 3c; Supplementary Figure S4).
According to sequence analysis from European samples, the dominant allele from mk245 (with six GTT repeats) was present in L. l. lepida individuals only. These results corroborate the genotypes obtained by Nunes et al. (2011a) for AFLP marker 245, as it was absent in populations from both ends of the environmental gradient (ALM and GAL) and reached higher frequencies in populations from the south of the Iberian Peninsula (CMA=0.80, TOL=0.95) (Figure 2). As for the African ocellated lizards, we detected four dominant-allele haplotypes (with six GTT repeats) and five recessive-allele haplotypes. Once more, the sample from Morocco that had European-like alleles for both mk75 and mk209 also had an European allele for mk245 (Figure 3c), and was the only African sample with <6 GTT repeats. Marker mk245 was also amplified in L. schreiberi, L. agilis and Iberolacerta monticola with success (Supplementary Figure S4).
Tajimas’ D values calculated to test for neutrality in each marker and each species or subspecies ranged from −1.48 to 1.82, but none differed significantly from zero (Table 2).
Data from sequenced samples were confronted with their respective AFLP electrophoresis profiles for markers mk75, mk209, mk245, mk386 and mk390 to identify bands that could correspond to the expected size of the recessive alleles. No AFLP markers with the expected size for the recessive allele could be found in mk75 or mk390 profiles. For markers mk209, mk245 and mk386, electrophoresis profiles presented AFLP fragments that matched with the predicted size of recessive alleles but their presence or absence in each sample was not always in agreement with the scoring expectations based on sequenced haplotypes, thus suggesting that they might have comigrated with non-homologous fragments of the same size (Supplementary Figure S5).
Discussion
AFLP outlier isolation and characterization
This study reports the follow-up of an AFLP genome scan in ocellated lizards performed by CE. Our attempts to isolate and sequence specific outliers from AFLP profiles and to convert them into codominant markers highlighted some pitfalls and technical challenges that are probably common to similar follow-up studies. We isolated outlier AFLP bands from agarose gels, but other higher resolution alternatives have been used to isolate outlier AFLPs, such as polyacrylamide (Midamegbe et al., 2011) or Spreadex gels (Minder and Widmer, 2008). Agarose gels were chosen for this study because it was the easiest and the most affordable solution to separate dense AFLP profiles, using CE to confirm the sizes of fragments recovered from gel slices and the sizes of clone inserts.
The first observation from this follow-up study was that isolation of small fragments (<150 bp) seems to be less successful because the fragment-size distribution in AFLP profiles is asymmetrical, with higher density of small-sized fragments. Additionally, it has been suggested that the chances of homoplasy are higher for smaller fragments (Vekemans et al., 2002; Caballero et al., 2008). Brugmans et al. (2003) proposed an alternative method for AFLP markers isolation, which reduces the density of bands in AFLP profiles before the excision of the desired band, without coisolation of adjacent fragments. This approach consists of the use of 12 MseI degenerate primers to determine, by subtractive amplification, the fourth, the fifth and the sixth selective base following the MseI restriction site. The band is then amplified with the appropriate MseI primer with six selective bases, excised from the gel, reamplified and used for direct sequencing. This method may greatly improve the efficiency of fragment recovery from gels, even for smaller fragments. Nevertheless, small fragments still impose some other limitations. The conversion of AFLP outliers into codominant loci might be compromised if no specific primers can be designed from the fragment sequence. In such cases, the known sequences of small-sized outliers must be expanded to their flanking regions through genome-walking strategies or homology with genomic databases in order to find suitable regions for primer design.
None of the seven outliers sequenced in ocellated lizards seem to belong to a coding region, but this is not a surprising observation. Mutational constraints in coding regions and the small size of AFLP markers (<500 bp) imply that most polymorphic AFLP markers probably fall in non-coding regions, which comprise most of the genome. Consequently, AFLP markers with outlier behaviour will rather be in close linkage with the gene under selection than inside the gene sequence itself (Stinchcombe and Hoekstra, 2008; Butlin, 2010) or act as regulatory elements. In accordance with these predictions, follow-up studies of AFLP genome scans available in the literature detected mostly non-coding outlier fragments, often including repetitive or transposable elements (Minder and Widmer, 2008; Wood et al., 2008; Paris and Despres, 2012).
With the knowledge of AFLP allele sequences, outliers can be converted in codominant markers with the development of specific primers, thus overcoming the disadvantages that dominant markers pose for population genomic analyses. A recent study by Foll et al. (2010) presented new developments on AFLP scoring methodology that account for band intensity in CE to genotype bands as codominant markers. This approach opens new perspectives on AFLP usefulness in population genomics, but the method depends on the use of high-quality electrophoresis profiles, because band intensity might be highly variable among samples because of technical variance in the generation of band profiles. Moreover, if the sequence content of AFLPs becomes available, it enables the screening of allelic frequencies through SNP genotyping, overcoming other AFLP weaknesses besides dominance, such as homoplasy or limitations in the transferability of AFLP markers to closely-related taxa.
Sources of length polymorphism between dominant and recessive alleles can be accessed from digested DNA with the combined use of outlier-specific internal primers and AFLP selective primers. In ocellated lizards, the sources of length polymorphism in investigated outliers were mostly internal indels (mk75, mk209 and mk386) or polymorphic microsatellites (mk245). The failure of the amplification of the full-fragment sequence for recessive alleles in mk75 could be caused by the unsuitability of the combination of outlier-specific and AFLP selective primers. However, mutations at recognition sites of one or both restriction enzymes are the most likely explanation and the extension of the outlier sequence toward its flanking regions by genome walking is necessary to access such sources of polymorphism.
Another observation from our ocellated lizard genome scan follow-up is that outlier fragments may often include one or more internal repetitive elements of variable length (for example, mk209 and mk245), thus resulting in several length-polymorphic alleles for the same locus, in the same population and generated by the same selective primer combination. If the repetitive element is formed by mononucleotide units (for example, G repeat in mk209), alleles with size differing in a single base will be very difficult to score unambiguously in AFLP electrophoresis profiles. If, however, the AFLP marker contains a microsatellite composed of trinucleotide units as for mk245, several band sizes are expected within the same population and they might bias AFLP band scores because of homoplasy, when allele size coincides with other non-homologous markers, or might lead to the non-independence of some polymorphic markers, if alleles of different size but from the same locus are scored as independent markers.
The risk for statistical bias caused by homoplasy and non-independence of anonymous markers like AFLPs has been noted before (Bonin et al., 2007, Caballero et al., 2008). Species with larger genomes are richer in repetitive elements and transposable elements. These regions have fewer mutational constraints and because of that are prone to contain AFLP restriction sites and generate several fragments of variable sizes. It has been suggested that smaller AFLP fragments (<150 bp) should be avoided to minimize homoplasy (Bonin et al., 2007). However, fragments with repetitive elements can easily achieve larger sizes, as in mk209 (303 bp) or mk245 (250 bp). Although some AFLP genome scan studies have tested for linkage disequilibrium among AFLP markers to control for non-independent markers (Murray and Hare, 2006; Paris et al., 2010; Poncet et al., 2010; Midamegbe et al., 2011; Paris and Despres, 2012), it might be difficult to detect linkage disequilibrium when the same AFLP locus generates >2 length-variable alleles potentially classified as different markers. Therefore, interpretation of results from AFLP outlier detection should be made with caution and special efforts should be taken to investigate the sequence content of outliers in order to validate their selection signature (Butlin, 2010).
Outlier sequence variation in ocellated lizards
Data from mk75 sequences revealed that the dominant band detected under selection in the genome scan corresponds to a quite conserved allele with a deletion of 9 bp that reaches higher frequencies in L. l. iberica. Although additional sources of length polymorphism might affect length polymorphism of mk75, the presence or absence of the internal deletion in sequenced samples is consistent with AFLP scoring frequencies from the genome scan. The dominant-allele sequence was also found in heterozygous samples from L. l. lepida, but it was not detected in L. l. nevadensis. Locus 75 was highlighted as an outlier only by BayeScan and its band presence was found in association with higher levels of precipitation, as registered in the northwest of the Iberian Peninsula, where L. l. iberica is found (Nunes et al., 2011a). Ocellated lizards normally cease their daily activities and remain in shelters when it rains. Thus, the smaller size and darker coloration of L. l. iberica are expected to be advantageous for thermoregulation efficiency in a rainy and colder environment and mk75 might be associated with these phenotypic traits. However, genomic resources available from reptile species in GenBank were not sufficient to find homology with the non-coding sequence of mk75 alleles, and therefore, the reason for the selection signature and possible effects on phenotype remain unknown and need further investigation.
For mk209, the TGGA indel justifies the length polymorphism of outlier 209. Sequence data indicate that the presence of TGGA is fixed in L. l. nevadensis, but absent in all other European ocellated lizards. Locus 209 was detected as an outlier only by DFDIST and its band presence was found in association with low levels of precipitation, as found in the southeast of the Iberian Peninsula, where L. l. nevadensis lives (Nunes et al., 2011a). The TGGA element is also present in most samples from African ocellated lizards, but their haplotypes are not conserved because of variation in repetitive elements. Because the neutral divergence of L. l. nevandensis is deep (Paulo et al., 2008; Miraldo et al., 2011), the role of drift might have been relevant for the fixation of mk209 in L. l. nevandensis, but the role of selection cannot be excluded with the current knowledge. This subspecies inhabits a region of reduced and irregular rainfall (<300 mm per year) and higher temperatures, leading to landscapes with sparse shrub-like vegetation. The different coloration of L. l. nevandensis seems efficient for camouflage in such landscapes (Nunes et al., 2011b) and the ability of females to lay >1 annual clutch with extended laying periods has been considered as an adaptive response to the irregular rainfall in the region (Mateo and Castanet, 1994). Nevertheless, more genomic resources are needed to understand which genes might be linked with locus mk209 and its importance in L. l. nevadensis evolution.
Sequence data from mk245 are perhaps the most surprising. A microsatellite composed of GTT repeats is responsible for locus 245 length polymorphism. Locus 245 was detected as an outlier by both DFDIST and BayeScan, and was the one with the strongest association with maximum temperature variation along the Iberian Peninsula (Nunes et al., 2011a). The dominant allele corresponds to six repeats of GTT and is absent in L. l. iberica and L. l. nevadensis samples, as expected from band frequency scored for locus 245 in the genome scan. The most striking observation is that mk245 alleles found in L. l. iberica and L. l. nevadensis have all the three GTT repeats, but the ecological settings faced by these subspecies are actually the most contrasted ones across the species range. Sequences of mk245 in samples from L. l. lepida populations are polymorphic and show not only the dominant allele, but also alleles with 3–5 GTT repeats. If locus mk245 is linked with genes that are being selected to respond to higher temperatures, then we would not expect that L. l. nevadensis would have the same fixed allele as L. l. iberica, which lives in a much colder region.
Microsatellites detected in loci mk209 and mk245 were even more variable in ocellated lizards from North Africa, but their flanking regions remain quite conserved across species. Haplotypes found in L. pater and L. tangitana for the three investigated loci (mk75, mk209 and mk245) are normally distinct from the ones found in European ocellated lizards, with one remarkable exception: a single individual from Morocco (L. tangitana) carries European haplotypes for the three loci. The same individual carries also an European allele at the melanocortin-1 receptor gene (JF732954, Nunes et al., 2011b). Although technical errors cannot be entirely ruled out, the cytochrome b haplotype obtained for this individual undoubtedly belongs to the North African clade (AF378959, Paulo et al., 2008), and it clusters with Moroccan samples based on neutral microsatellites (Paulo, unpublished data). Given the long time (about 11 million years) of divergence and isolation of this species from European lizards (Paulo et al., 2008), it is unexpected to find such a level of ancestral polymorphism retained in a single individual at the nuclear level. Perhaps this might be indicative of past episodes of secondary contact between African and European lizards before the full separation of both continents with the opening of the Strait of Gibraltar at the end of the Messinian salinity crisis (5.33 million years ago), but it remains extremely speculative with the current knowledge on African ocellated lizards.
Future directions
The sequence data obtained so far for the investigated outliers is restricted, and neither confirms nor denies the existing evidence that they might be candidates influenced by selection. None of them correspond to coding DNA, implying that they may be involved either in the regulation of genes under selection or simply be in linkage disequilibrium with the actual target of selection. Our sequence data do not point to duplications of loci, but this is a scenario that must be considered when characterizing outlier loci, especially if transposable elements are involved, as their sequences can be repeated in several locations of the genome (for example, Wood et al., 2008; Paris and Despres, 2012). Thus, in order to proceed with the characterization of outlier loci in non-model species such as L. lepida, it is necessary to develop additional and extensive genomic resources.
The data obtained so far in ocellated lizards from outlier sequences of dominant and recessive alleles showed that these loci represent small regions in the genome that diverge substantially between L. lepida subspecies. L. l. nevadensis has been evolving independently for a long time, accumulating a deep divergence that probably results from both natural selection and the accumulation of neutral divergence. In the opposite end of the environmental gradient, L. l. iberica presents a considerable divergence based on outlier loci but not in neutral loci, suggesting an incipient stage of putative ecological speciation. Future investigation in ocellated lizards should focus in the genetic variation at the contact zones between subspecies to validate these hypotheses about their evolutionary history.
DATA archiving
Sequence data have been deposited at GenBank: accession numbers JQ268560-JQ268566 and JQ310676-JQ310742.
Accession codes
References
Apple JL, Grace T, Joern A, Amand PS, Wisely SM (2010). Comparative genome scan detects host-related divergent selection in the grasshopper Hesperotettix viridis. Mol Ecol 19: 4012–4028.
Bandelt H-J, Forster P, Röhl A (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48.
Beaumont MA, Nichols RA (1996). Evaluating loci for use in the genetic analysis of population structure. Proc R Soc Lond B Biol Sci 263: 1619–1626.
Bensch S, Akesson M (2005). Ten years of AFLPs in ecology and evolution: why so few animals? Mol Ecol 14: 2899–2914.
Bonin A, Ehrich D, Manel S (2007). Statistical analysis of amplified fragment length polymorphism data: a toolbox for molecular ecologists and evolutionists. Mol Ecol 16: 3737–3758.
Bonin A, Taberlet P, Miaud C, Pompanon F (2006). Explorative genome scan to detect candidate loci for adaptation along a gradient of altitude in the common frog (Rana temporaria). Mol Biol Evol 23: 773–783.
Brugmans B, van der Hulst RGM, Visser RGF, Lindhout P, van Eck HJ (2003). A new and versatile method for the successful conversion of AFLP markers into simple single locus markers. Nucleic Acids Res 31: e55.
Butlin RK (2010). Population genomics and speciation. Genetica 138: 409–418.
Caballero A, Quesada H, Rolan-Alvarez E (2008). Impact of amplified fragment length polymorphism size homoplasy on the estimation of population genetic diversity and the detection of selective loci. Genetics 179: 539–554.
Excoffier L, Hofer T, Foll M (2009). Detecting loci under selection in a hierarchically structured population. Heredity 103: 285–298.
Excoffier L, Laval G, Schneider S (2005). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47–50.
Flot JF, Tillier A, Samadi S, Tillier S (2006). Phase determination from direct sequencing of length-variable DNA regions. Mol Ecol Notes 6: 627–630.
Foll M, Fischer MC, Heckel G, Excoffier L (2010). Estimating population structure from AFLP amplification intensity. Mol Ecol 19: 4638–4647.
Foll M, Gaggiotti O (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180: 977–993.
Hall TA (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41: 95–98.
Joost S, Kalbermatten M, Bonin A (2008). Spatial analysis method (SAM): a software tool combining molecular and environmental data to identify candidate loci for selection. Mol Ecol Resour 8: 957–960.
Mateo JA, Castanet J (1994). Reproductive strategies in 3 Spanish populations of the ocellated lizard, Lacerta lepida (Sauria, Lacertidae). Acta Oecol 15: 215–229.
Mateo JA, Castroviejo J (1990). Variation morphologique et révision taxonomique de l'espèce Lacerta lepida Daudin, 1802 (Sauria, Lacertidae). Bull Mus Natl Hist Nat, Paris 12: 691–706.
Meksem K, Ruben E, Hyten D, Triwitayakorn K, Lightfoot DA (2001). Conversion of AFLP bands into high-throughput DNA markers. Mol Genet Genomics 265: 207–214.
Midamegbe A, Vitalis R, Malausa T, Delava E, Cros-Arteil S, Streiff R (2011). Scanning the European corn borer (Ostrinia spp.) genome for adaptive divergence between host-affiliated sibling species. Mol Ecol 20: 1414–1430.
Minder AM, Widmer A (2008). A population genomic analysis of species boundaries: neutral processes, adaptive divergence and introgression between two hybridizing plant species. Mol Ecol 17: 1552–1563.
Miraldo A, Hewitt GM, Paulo OS, Emerson BC (2011). Phylogeography and demographic history of Lacerta lepida in the Iberian Peninsula: multiple refugia, range expansions and secondary contact zones. BMC Evol Biol 11: 170.
Murray MC, Hare MP (2006). A genomic scan for divergent selection in a secondary contact zone between Atlantic and Gulf of Mexico oysters, Crassostrea virginica. Mol Ecol 15: 4229–4242.
Nunes VL, Beaumont MA, Butlin RK, Paulo OS (2011a). Multiple approaches to detect outliers in a genome scan for selection in ocellated lizards (Lacerta lepida) along an environmental gradient. Mol Ecol 20: 193–205.
Nunes VL, Miraldo A, Beaumont MA, Butlin RK, Paulo OS (2011b). Association of Mc1r variants with ecologically relevant phenotypes in the European ocellated lizard, Lacerta lepida. J Evol Biol 24: 2289–2298.
Paris M, Boyer S, Bonin A, Collado A, Jean-Philippe D, Despres L (2010). Genome scan in the mosquito Aedes rusticus: population structure and detection of positive selection after insecticide treatment. Mol Ecol 19: 325–337.
Paris M, Despres L (2012). Identifying insecticide resistance genes in mosquito by combining AFLP genome scans and 454 pyrosequencing. Mol Ecol 21: 1672–1686.
Paulo OS, Pinheiro J, Miraldo A, Bruford MW, Jordan WC, Nichols RA (2008). The role of vicariance vs. dispersal in shaping genetic patterns in ocellated lizard species in the western Mediterranean. Mol Ecol 17: 1535–1551.
Pérez-Figueroa A, García-Pereira M, Saura M, Rolán-Alvarez E, Caballero A (2010). Comparing three different methods to detect selective loci using dominant markers. J Evol Biol 23: 2267–2276.
Pina-Martins F, Paulo OS (2008). Concatenator: sequence data matrices handling made easy. Mol Ecol Resour 8: 1254–1255.
Polanco C, González AI, de la Puente R, Somalo S, Ruiz ML (2005). Cloning of AFLP markers detected by fluorescence capillary electrophoresis. Plant Mol Biol Rep 23: 271–277.
Poncet BN, Herrmann D, Gugerli F, Taberlet P, Holderegger R, Gielly L et al. (2010). Tracking genes of ecological relevance using a genome scan in two independent regional population samples of Arabis alpina. Mol Ecol 19: 2896–2907.
Reddy PS, Mahanty S, Kaul T, Nair S, Sopory SK, Reddy MK (2008). A high-throughput genome-walking method and its use for cloning unknown flanking sequences. Anal Biochem 381: 248–253.
Rowe HC, Renaut S, Guggisberg A (2011). RAD in the realm of next-generation sequencing technologies. Mol Ecol 20: 3499–3502.
Rozen S, Skaletsky HJ (2000). Primer 3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, (eds). Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana Press: Totowa, New Jersey. pp 365–386.
Stephens M, Scheet P (2005). Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet 76: 449–462.
Stephens M, Smith NJ, Donnelly P (2001). A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68: 978–989.
Stinchcombe JR, Hoekstra HE (2008). Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity 100: 158–170.
Tajima F (1989). Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Thompson JD, Higgins DG, Gibson TJ (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680.
Vekemans X, Beauwens T, Lemaire M, Roldán-Ruiz I (2002). Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Mol Ecol 11: 139–151.
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M et al. (1995). AFLP - a new technique for DNA-fingerprinting. Nucleic Acids Res 23: 4407–4414.
Wood HM, Grahame JW, Humphray S, Rogers J, Butlin RK (2008). Sequence differentiation in regions identified by a genome scan for local adaptation. Mol Ecol 17: 3123–3135.
Wong A, Forbes MR, Smith ML (2001). Characterization of AFLP markers in damselflies: prevalence of codominant markers and implications for population genetic applications. Genome 44: 677–684.
Acknowledgements
This research was supported by a doctoral scholarship from FCT (SFRH/BD/21306/2005) to Nunes VL and the FCT project POCI/BIA/59288/2004—‘Process of speciation: accessing the genic view’, coordinated by Paulo OS. We thank three anonymous reviewers for their helpful comments on a previous version of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Rights and permissions
About this article
Cite this article
Nunes, V., Beaumont, M., Butlin, R. et al. Challenges and pitfalls in the characterization of anonymous outlier AFLP markers in non-model species: lessons from an ocellated lizard genome scan. Heredity 109, 340–348 (2012). https://doi.org/10.1038/hdy.2012.48
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2012.48
Keywords
This article is cited by
-
Geropogon hybridus (L.) Sch.Bip. (Asteraceae) exhibits micro-geographic genetic divergence at ecological range limits along a steep precipitation gradient
Plant Systematics and Evolution (2017)
